05 March 2021: Database Analysis
Identification of NDRG Family Member 4 (NDRG4) and CDC28 Protein Kinase Regulatory Subunit 2 (CKS2) as Key Prognostic Genes in Adrenocortical Carcinoma by Transcriptomic Analysis
Zhengqing Yang1BE, Hui Cheng1CD, Yazhou Zhang1F, Yan Zhou1AG*DOI: 10.12659/MSM.928523
Med Sci Monit 2021; 27:e928523






Abstract
BACKGROUND: Adrenocortical carcinoma (ACC) is an aggressive cancer with heterogeneous outcomes. In this study, we aimed to investigate genomic and prognostic features of ACC.
MATERIAL AND METHODS: Clinical, pathologic, and transcriptomic data from 2 independent datasets derived from ACC samples (TCGA-ACC dataset, GEO-GSE76021 dataset) were collected. Weighted gene co-expression network analysis (WGCNA) and survival analyses were performed to identify prognostic genes. Pathway analysis was performed for mechanistic analysis. xCell deconvolution was performed for tumor microenvironment analysis.
RESULTS: In the TCGA-ACC cohort, WGCNA identified a prognostic module of 5408 genes. Differential expression analysis identified 1969 genes that differed in expression level between long-term and short-term survivors. Univariate Cox regression model analysis identified 8393 genes with prognostic value. The intersection of these gene sets included 820 prognostic genes. Similar protocols were performed for the GSE76021 dataset, and 5 candidate genes were identified. Further intersection of these genes finally identified NDRG4 and CKS2 as key prognostic genes. Multivariate Cox regression model analysis validated the prognostic value of NDRG4 (HR=0.61, 95% CI 0.46–0.80) and CKS2 (HR=2.52, 95% CI 1.38–4.60). Moreover, NDRG4 and CKS2 expression predicted survival in patients treated with mitotane (P<0.001). Further mechanism exploration found an association between CKS2 and DNA mismatch repair pathways. Moreover, NDRG4 positively correlated with CD8⁺ T cell infiltration, while CKS2 negatively correlated with it.
CONCLUSIONS: We identified NDRG4 and CKS2 expression as key prognostic genes in ACC, which may help in risk stratification of ACC. Moreover, a close relationship was found between CKS2 and mismatch repair pathways. Moreover, immune cell infiltration differed according to NDRG4 and CKS2 expression.
Keywords: adrenocortical carcinoma, Biological Markers, Adrenal Cortex Neoplasms, Biomarkers, Tumor, CDC2-CDC28 Kinases, Cell Cycle Proteins, Gene Expression Profiling, Muscle Proteins, Nerve Tissue Proteins, Proportional Hazards Models, Protein Kinases, tumor microenvironment
Background
Adrenocortical carcinoma (ACC) is an aggressive cancer originating in the cortex of the adrenal gland [1]. Almost half of ACC patients will experience recurrence or metastasis [2], resulting in a relatively low 5-year overall survival (OS) rate, ranging from 16% to 60% [3,4]. Traditional prognostic factors for ACC include tumor stage [5], resection status [6], and Ki67 index of tumor proliferation [7]. However, these traditional factors still have limitations. Recent studies have identified novel molecular biomarkers for predicting ACC survival, such as a 5-gene (TOP2A, NDC80, CEP55, CDKN3, and CDK1) signature, as well as DNA damage and cell cycle pathways [8–12], indicating that the molecular pathogenesis of ACC may also be involved in disease progression and survival.
Advances in molecular biology have uncovered part of the pathogenesis mechanism of ACC [13–17], but the precise mechanisms are still far from known. Recently, genomic approaches have provided a more comprehensive landscape for ACC pathogenesis [18,19]. Assié et al were the first to apply exome sequencing and SNP array analysis in a relatively large number of ACC patients [19]. They identified 2 distinct molecular ACC subgroups with opposite outcome [19]. Another study by Zheng et al applied a comprehensive pan-genomic analysis of ACC patients, which identified PRKAR1A, RPL22, TERF2, CCNE1, and NF1 as new driver genes of ACC [18]. They also divided ACC into 3 subtypes according to a DNA-methylation signature, which may be helpful in the risk stratification of ACC [18]. These high-throughput genomic studies helped to expand knowledge boundaries with regard to ACC development.
Mitotane (1,1-(dichlorodiphenyl)-2,2-dichloroethane) is a steroidogenesis inhibitor and cytostatic antineoplastic medication for ACC treatment, which is recommended as systemic therapy for metastatic or unresectable ACC, as well as adjuvant therapy for high-risk postoperative patients [20]. However, not all patients respond to mitotane. Recently, the pharmacokinetics of mitotane has been found to be a predictive factor in ACC prognosis [21,22]. A recent study also identified the correlation between SOAT1 expression and response to mitotane in ACC patients [23]. However, no robust predictive biomarker has been applied in the clinic.
In the present study, by re-evaluation of 2 open-access transcriptomics datasets, we aimed to identify key prognostic genes in ACC, which may be predictive of the efficacy of mitotane therapy as well. Moreover, we attempted to discover the molecular mechanisms underlying the functions of these prognostic genes.
Material and Methods
STUDY POPULATIONS AND DATA ACQUISITION:
The study design is illustrated in Figure 1. Two independent datasets, the TCGA-ACC dataset and the GEO-GSE76021 dataset, were downloaded for the study. The TCGA-ACC cohort included 79 patients (Table 1) and the GEO-GSE76021 cohort included 29 patients. For the TCGA-ACC cohort, clinical, pathologic, and transcriptomic data were downloaded from the UCSC Xena (https://xena.ucsc.edu/) website. For the GEO-GSE76021 cohort, clinical, pathologic, and transcriptomic data were downloaded from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) website. R software (version 4.0.2; https://www.r-project.org/) and Bioconductor packages (http://www.bioconductor.org/) were used for data analysis. The “sva” package [24] was used for the normalization of the GSE76021 dataset. For duplicates, RNA expression was averaged, and genes with relatively low abundance were discarded. We followed the access policies of the TCGA and GEO databases strictly during the process of the study.
WEIGHTED GENE CO-EXPRESSION NETWORK ANALYSIS:
Weighted gene co-expression network analysis (WGCNA) was used as a systematic method to describe the pattern of gene correlation, as previously described by Langfelder et al [25]. We set OS as the phenotype for WGCNA analysis. Potential biomarker genes in ACC were identified through association between gene sets and OS. In the current study, gene expression matrices for the TCGA-ACC cohort and the GEO-GSE76021 cohort were used as input data. The “WGCNA” package in R software was used for WGCNA processing. An adjacency matrix and a topological overlap matrix were constructed. After that, we constructed a gene dendrogram, and performed module identification with a dynamic tree cut. The correlation between OS and genomic data was then calculated. The most predominant module associated with OS was selected as the key module, and genes in the key module were used in the following analysis.
SURVIVAL ANALYSIS:
Univariate Cox analysis was used to screen survival-related genes. Hazard ratio (HR) was used to classify genes as either protective or deleterious. These genes were selected as potential prognostic genes. A multivariate Cox regression model including gender, age, tumor stage, NDRG Family Member 4 (NDRG4), and CDC28 Protein Kinase Regulatory Subunit 2 (CKS2) was used to identify independent prognostic factors for OS in ACC. Kaplan-Meier survival analysis and log-rank test were used to validate the prognostic value of target genes.
DIFFERENTIAL EXPRESSION ANALYSIS:
For differential expression analysis, long-term survivors were defined as those with OS >5 years, and short-term survivors were defined as those with OS <3 years. Differentially expressed genes (DEGs) were determined by the “Limma” package moderated t test, and genes with P value <0.001 and fold change >2 were regarded as DEGs [26].
ESTABLISHMENT OF REGRESSION MODEL AND CONSTRUCTION OF RISK SCORE:
The risk score for predicting postoperative survival was calculated based on a linear combination of tumor stage and gene expression in the TCGA-ACC cohort by logistic regression. The following calculation formula was used for the analysis:
N, Coei, and Expi represented gene number, coefficient value, and level of gene expression, respectively. Receiver operating characteristic (ROC) analysis was used to evaluate the accuracy of the prognostic model.
GENE SET ENRICHMENT ANALYSIS:
Gene set enrichment analysis (GSEA) was performed using the molecular signatures database (MSigDB) (
TUMOR MICROENVIRONMENT ANALYSIS:
We applied xCell (http://xCell.ucsf.edu/), a gene signature-based deconvolution method, to infer 64 immune and stromal cell types for ACC microenvironment analysis [27].
STATISTICAL ANALYSIS:
Wilcoxon signed ranks test or
Results
IDENTIFICATION OF POTENTIAL PROGNOSTIC GENES IN THE TCGA-ACC COHORT:
Figure 1 shows the study design. We first performed weighted correlation network analysis to identify prognostic genes in the TCGA-ACC cohort. OS was set as the phenotype. Expression matrices for 79 samples were used for WGCNA construction. Figure 2A shows the cluster dendrogram of the samples in the TCGA-ACC cohort. After quality assessment for each expression matrix, R2=0.9 was selected to ensure a scale-free network (Figure 2B). We also set a threshold of 0.4 to merge similar modules. After that, a total of 15 modules were identified (Figure 2C). Genes in the grey module were removed in the subsequent analysis. Next, we calculated eigengenes for all of the modules and clustered them based on their correlation (Figure 2D, 2E). A module eigengene heatmap showed that the 7 modules were mainly divided into 3 clusters (Figure 2D). A network heatmap was also performed to analyze the interactions between the modules (Figure 2E). The results showed that the skyblue2 module was relatively independent from the other modules, with a high-scale independence of transcriptomic expression. Figure 2F shows a heatmap of the correlation between module eigengenes and patients’ survival in the TCGA-ACC cohort. Close relationships are represented by red color. We found that the skyblue2 module was significantly correlated with OS (Figure 2F, P=9×10−8). Taken together, the skyblue2 module was selected as the main prognostic module.
We further performed differential expression analysis to define 3769 DEGs between long-term survivors (OS >5 years) and short-term survivors (OS <3 years) in the TCGA-ACC cohort (Figure 2G). Moreover, we performed a univariate Cox regression model to define 8393 potential prognostic genes (Figure 2H). Finally, 820 intersection genes were defined between the DEGs, Cox regression prognostic genes, and the WGCNA-skyblue2 module genes.
IDENTIFICATION OF POTENTIAL PROGNOSTIC GENES IN THE GSE76021 COHORT:
We downloaded clinicopathologic and transcriptomic data from the GEO dataset GSE76021, which consisted of data from 29 ACC patients. We also set OS as the phenotype for WGCNA. A cluster dendrogram of the samples in the GSE76021 cohort is illustrated in Figure 3A. After quality assessment for the expression matrix, R2=0.9 was selected to ensure a scale-free network (Figure 3B). We also set a threshold of 0.4 to merge similar modules. After that, a total of 16 modules were identified (Figure 3C). Next, we calculated eigengenes for all modules and clustered them based on their correlation (Figure 3D, 3E). The modules were mainly divided into 2 clusters (Figure 3D), and the interactions among all of the modules were detected by network heatmap (Figure 3E). In the end, the plum3 module was defined as relatively independent from the other modules, with a high-scale independence of transcriptomic expression. Moreover, a close relationship was found between the plum3 module and the patients’ OS in the GSE76021 cohort (Figure 3F). Taken together, the plum3 module was selected as the main prognostic module in the GSE76021 cohort.
Differential expression analysis was further performed in the GSE76021 cohort, and 410 DEGs were found between long-term survivors (OS >5 years) and short-term survivors (OS <3 years) (Figure 3G). Moreover, univariate Cox analysis was performed, and 1059 potential prognostic genes were identified (Figure 3H). Finally, 5 intersection genes were defined between the DEGs, Cox regression prognostic genes, and WGCNA-plum3 module genes in the GSE76021 cohort.
IDENTIFICATION OF NDRG4 AND CKS2 AS PROGNOSTIC GENES FOR ACC:
Figure 4A shows a Venn diagram of the intersection of prognostic genes in the TCGA-ACC cohort and the GSE76021 cohort. We next constructed a multivariate Cox regression model for OS in ACC, including gender, age, tumor stage, NDRG4, and CKS2 (Figure 4B). In this multivariate Cox regression model, NDRG4 and CKS2 expression were both independently associated with the patients’ OS (NDRG4: HR 0.61, 95% CI 0.46–0.80, P<0.001; CKS2: HR 2.52, 95% CI 1.38–4.60, P=0.003; Figure 4B). Moreover, NDRG4 expression decreased when tumor stage increased (Figure 4C). In contrast, CKS2 expression increased when tumor stage increased. The results indicated that NDRG4 and CKS2 expression are potential prognostic genes in ACC, and are associated with ACC progression.
We also performed Kaplan-Meier survival analysis for the ACC patients. Kaplan-Meier survival curves and log-rank analysis showed that high expression of NDRG4 was associated with better survival in both the TCGA-ACC cohort (P<0.001; Figure 5A) and the GSE76021 cohort (P=0.002; Figure 5B). On the other hand, CKS2 high expression was associated with worse survival in both the TCGA-ACC cohort (P<0.001; Figure 5A) and the GSE76021 cohort (P<0.001; Figure 5B). For disease-free survival, Kaplan-Meier survival analysis also identified NDRG4 (P<0.001) and CKS2 (P<0.001) expression as prognostic factors (Figure 5C). Moreover, NDRG4 (P<0.001) and CKS2 (P<0.001) expression could also be prognostic factors for patients treated with mitotane therapy (Figure 5D).
We further performed ROC analysis to identify whether CKS2 and NDRG4 could be used as predictors for 3-year survival and 5-year survival of ACC patients. We constructed a combined risk score by logistic regression modelling for ACC survival according to tumor stage, NDRG4 expression, and CKS2 expression. We found that this combined risk score could be used for prediction of 3-year survival in ACC, and this worked better than tumor stage alone (AUC=0.974 vs AUC=0.804, P=0.0016, Figure 5E). The combined risk score could also be used for prediction of 5-year survival, a stronger prediction than for tumor stage alone (AUC=0.969 vs AUC=0.891, P=0.0313; Figure 5F).
BIOLOGIC PATHWAYS RELATED TO NDRG4 AND CKS2 EXPRESSION:
In order to clarify biologic pathways related to NDRG4 and CKS2 expression, we further performed GSEA analysis in the TCGA-ACC cohort. We found several pathways enriched in NDRG4 low-expression samples including the cell cycle pathway, the DNA replication pathway, the hedgehog signaling pathway, and the homologous recombination pathway (Figure 6A). Moreover, expression of genes related to the homologous recombination pathway was mostly elevated in the NDRG4 low-expression samples (Figure 6B). In addition, a negative correlation was found between NDRG4 expression and homologous recombination genes including BRCA1, BRCA2, EXO1, and MSH2 (Figure 6C).
We also performed GSEA analysis between low and high expression levels of CKS2. We found DNA replication mismatch repair, p53 signaling pathway, and spliceosome pathway genes were enriched in CKS2 high-expression samples (Figure 6D). Moreover, expression of genes related to DNA mismatch repair pathways was mostly elevated in the CKS2 high-expression samples (Figure 6E). In addition, a positive correlation was found between NDRG4 expression and mismatch repair genes such as BRCA1, BRCA2, EXO1, and MSH2 (Figure 6F).
IMMUNE CELL INFILTRATION ASSOCIATED WITH NDRG4 AND CKS2 EXPRESSION:
Immunotherapy has been recently applied in multiple cancer types. We aimed to discover whether NDRG4 and CKS2 expression was associated with immune cell infiltration in ACC. By using xCell cell type enrichment analysis, we identified immune cell infiltration in the TCGA-ACC samples. The infiltration of CD8+ T cells, dendritic cells, natural killer T cells, regulatory T cells, macrophages, and fibroblasts was significantly more abundant in NDRG4 high-expression samples, while the infiltration of Th1 and Th2 cells was more abundant in NDRG4 low-expression samples (Figure 7A). Correlation analysis also identified a positive correlation between NDRG4 expression and infiltration of CD8+ T cells, dendritic cells, natural killer T cells, regulatory T cells, macrophages, and fibroblasts; and a negative correlation between NDRG4 expression and infiltration of Th1 and Th2 cells (Figure 7B).
We also analyzed the relationship between CKS2 expression and immune cell infiltration. The infiltration of CD8+ T cells, dendritic cells, natural killer T cells, regulatory T cells, macrophages, and fibroblasts was significantly more abundant in CKS2 low-expression samples, while the infiltration of Th1 and Th2 cells was more abundant in CKS2 high-expression samples (Figure 7C). Correlation analysis also identified a negative correlation between CKS2 expression and infiltration of CD8+ T cells, dendritic cells, natural killer T cells, regulatory T cells, macrophages, and fibroblasts; and a positive correlation between CKS2 expression and infiltration of Th1 and Th2 cells (Figure 7D). Our results suggested immune cell infiltration differs according to NDRG4 and CKS2 expression.
Discussion
ACC is an aggressive cancer with heterogeneous outcomes, but its molecular mechanisms have not yet been clarified. Recently, genomic technologies have been applied for the discovery of mechanisms of ACC. In the present study, we applied transcriptional analysis of available transcriptomic data from 2 datasets of ACC. We identified NDRG4 and CKS2 as key prognostic genes for ACC, with potential predictive value for mitotane therapy as well. We further identified mismatch repair pathways as dominant biologic pathways associated with CKS2. Moreover, immune cell infiltration in ACC differed according to NDRG4 and CKS2 expression.
NDRG4 is a member of the N-myc downregulated gene family, which belongs to the alpha/beta hydrolase superfamily [28]. The protein encoded by this gene is a cytoplasmic protein that is required for cell cycle progression and survival [29], and may be involved in the regulation of mitogenic signaling in vascular smooth muscles cells [30]. Research into the connection between NDRG4 and cancer is gaining more and more attention, although conflicting results have been reported. NDRG4 can show either tumor-suppressive or oncogenic function, depending on the tumor types [31–34]. Currently, the function of NDRG4 in ACC has not been determined. Our study found a relationship between high expression of NDRG4 and favorable survival in ACC (Figure 5A–5C), suggesting that it may be a potential tumor suppressor. Since NDRG4 is required for tumor cell cycle regulation [29], and cell cycle regulation plays a crucial role in ACC progression [12], we believe this could be a potential mechanism underlying the correlation between NDRG4 and ACC development. Moreover, Agosta et al (2018) found that NDRG4 may be a potential target of miR-139-5p, and that the miR-139-5p/NDRG4 pathway promotes ACC aggressiveness by mediating epithelial-to-mesenchymal transition, which results in tumor cell invasiveness and anchorage-independent growth [35]. However, this finding needs further in vitro and in vivo validation.
CKS2 is a protein-coding gene that binds to the catalytic subunit of the cyclin-dependent kinases and is essential for their biological function [36]. CKS2 is involved in the process of tumorigenesis of gastric cancer [37], prostate cancer [38], bladder cancer [39] and others. Our study also found an association between high expression of CKS2 and poor survival in ACC (Figure 5A–5C). Moreover, we found CKS2 expression to be correlated with elevated expression of mismatch repair genes such as BRCA1, BRCA2, EXO1, and MSH2 (Figure 6D–6F). Since CKS2 overexpression has been found to be able to override the DNA damage response barrier triggered by activated oncoproteins [40], we believe this could be a potential mechanism underlying the correlation between CKS2 and survival of ACC patients. Moreover, through in vitro experiments, Chehade et al found that miR-7, which participates in cell cycle arrest in ACC, acts as a key regulator of CKS2 [41]. According to these studies, we speculate that miRNAs might play an important role in the regulation of NDRG4 and CKS2, resulting in tumor cell proliferation and aggressiveness, as well as ACC progression. However, further in vitro and in vivo functional studies are still needed. In vitro studies needed include gene overexpression and CRISPR knock-out in tumor cells, and evaluation of tumor cell growth, invasion, and other related biological processes. In vivo studies could be performed on tumor-bearing mice, including evaluation of tumor growth and mouse survival. Moreover, the functional status of immune cells in tumor-bearing mice should also be evaluated, since our study also suggested a relationship between CKS2/NDRG4 and intratumoral immunity.
Mitotane is recommended as systemic therapy for metastatic or unresectable ACC, and also as adjuvant therapy in high-risk postoperative patients [20], but not all patients respond to this drug. The development of robust predictive biomarkers for mitotane therapy efficacy may have potential clinical significance. Our study identified NDRG4 and CKS2 as predictive biomarkers for mitotane therapy success in ACC (Figure 5D), which may be helpful in clinical management. However, further validation studies in larger, prospective cohorts are still needed.
Sequence-based analyses, including genomic, transcriptomic, and epigenomic profiling, have been applied recently in ACC studies [18]. Such studies have identified novel driver genes and molecular classifications for ACC, which may guide further functional studies [18]. Our study integrated multiple datasets from previous studies, which provided a more comprehensive view of the pathogenesis of ACC.
The present study still has some limitations. The limited sample size is the major limitation. Further validation studies in larger, prospective cohorts are needed. Moreover, the methodology of the current study could not clarify the underlying molecular mechanism of the 2 prognostic genes. Future functional and mechanical studies are needed to fully understand the underlying mechanisms. In addition, the study indicated a relationship between immunogenic status and prognosis in ACC. Further studies are needed to clarify whether or not immune cell infiltration is involved in ACC pathogenesis and development, and whether immune cell infiltration could be a potential therapeutic target in ACC. The prognostic genes identified in this preliminary study require validation by further interventional and functional studies.
Conclusions
This study applied a comprehensive transcriptomic approach to analyze available transcriptomic data to identify key prognostic genes in ACC. We identified NDRG4 and CKS2 as key prognostic genes in ACC that also may have potential predictive value for mitotane therapy. We further identified DNA mismatch repair pathways as dominant biologic pathways associated with CKS2. Moreover, immune cell infiltration in ACC differed according to NDRG4 and CKS2 expression. These findings require validation by further interventional and functional studies.
Figures
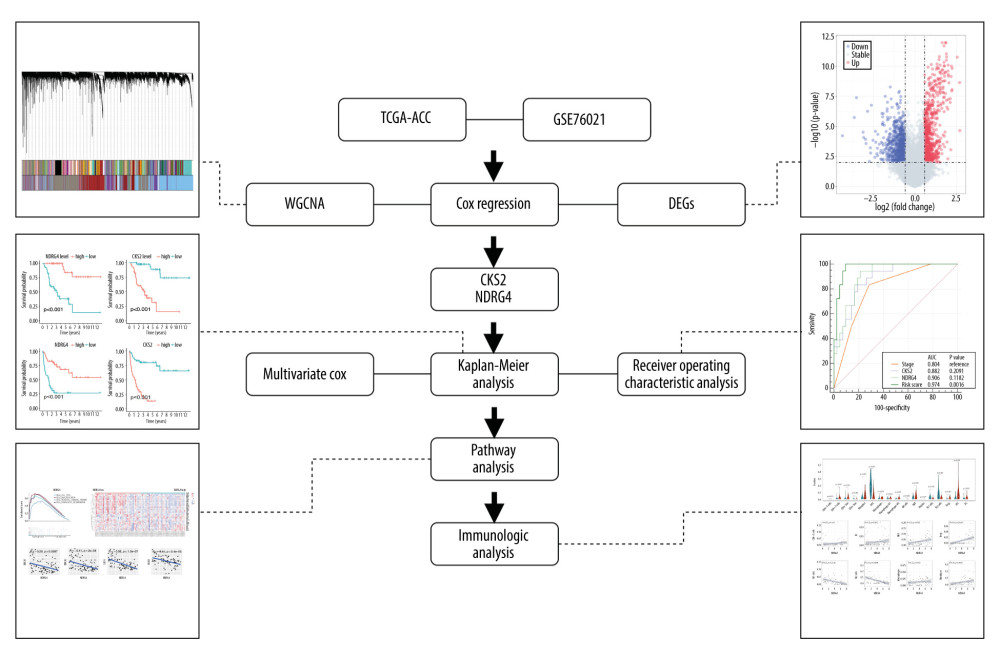 Figure 1. Flow chart of the study design.
Figure 1. Flow chart of the study design. 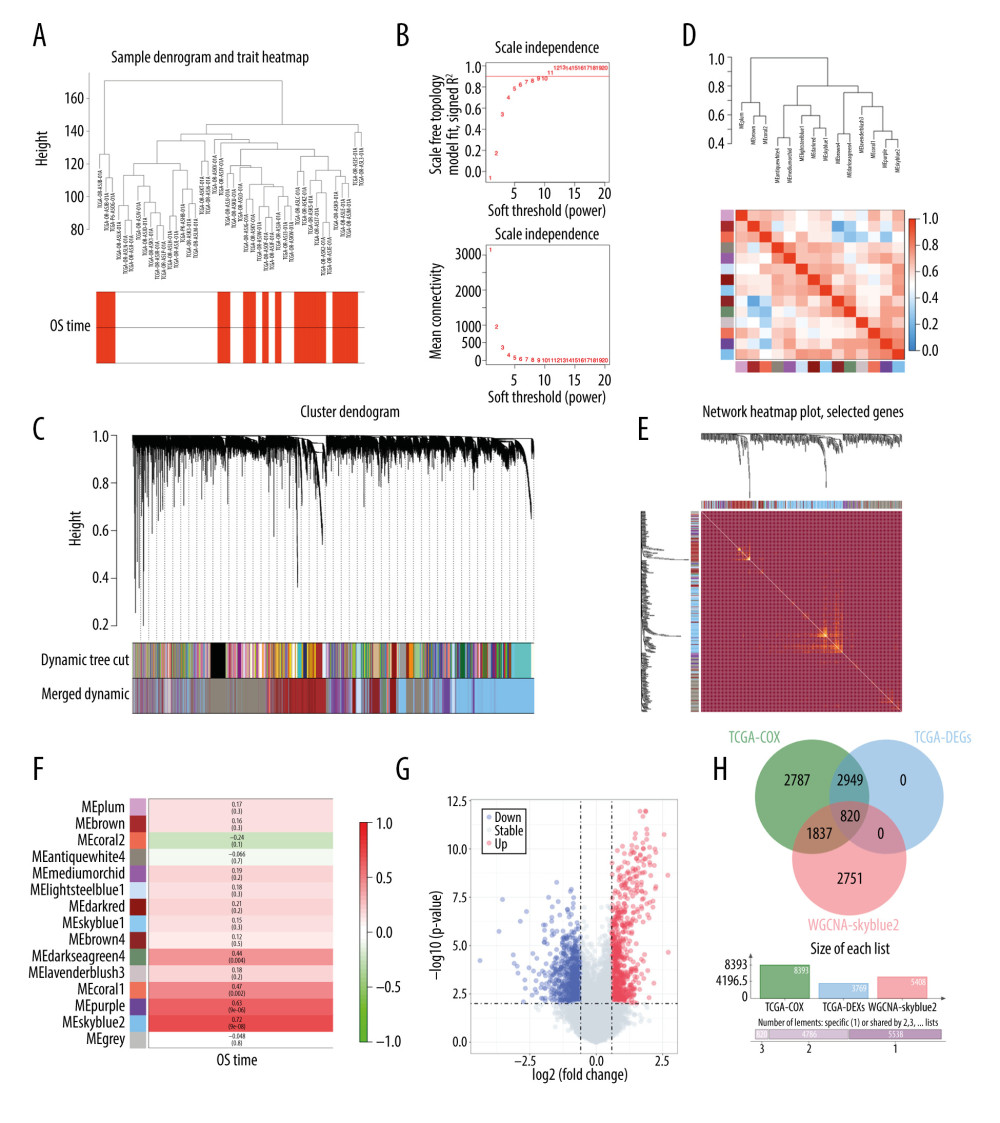 Figure 2. Identification of prognostic genes in the TCGA-ACC cohort by WGCNA. (A) Cluster dendrogram of samples in the TCGA-ACC cohort to detect outliers. The dendrogram branches represent the clustered samples. (B) Soft-threshold power analysis of the scale-free fit index (upper) and the mean connectivity (lower). (C) Dendrogram of gene clustering and module assignment. Modules are represented by different colors. (D) Heatmap of adjacencies in the eigengene network. Higher adjacency is represented by red color. (E) Interaction of co-expressing modules. Higher connectivity is represented by light color. (F) Heatmap of the correlation between module eigengenes and patients’ survival. Close relationships are represented by red color. (G) Volcano plot of DEGs between long-term survivors and short-term survivors in the TCGA-ACC cohort. (H) Venn diagram of the intersection of DEGs between long-term survivors and short-term survivors, prognostic genes according to univariate Cox regression model, and the WGCNA-skyblue2 module. WGCNA – weighted correlation network analysis; DEGs – differentially expressed genes.
Figure 2. Identification of prognostic genes in the TCGA-ACC cohort by WGCNA. (A) Cluster dendrogram of samples in the TCGA-ACC cohort to detect outliers. The dendrogram branches represent the clustered samples. (B) Soft-threshold power analysis of the scale-free fit index (upper) and the mean connectivity (lower). (C) Dendrogram of gene clustering and module assignment. Modules are represented by different colors. (D) Heatmap of adjacencies in the eigengene network. Higher adjacency is represented by red color. (E) Interaction of co-expressing modules. Higher connectivity is represented by light color. (F) Heatmap of the correlation between module eigengenes and patients’ survival. Close relationships are represented by red color. (G) Volcano plot of DEGs between long-term survivors and short-term survivors in the TCGA-ACC cohort. (H) Venn diagram of the intersection of DEGs between long-term survivors and short-term survivors, prognostic genes according to univariate Cox regression model, and the WGCNA-skyblue2 module. WGCNA – weighted correlation network analysis; DEGs – differentially expressed genes. 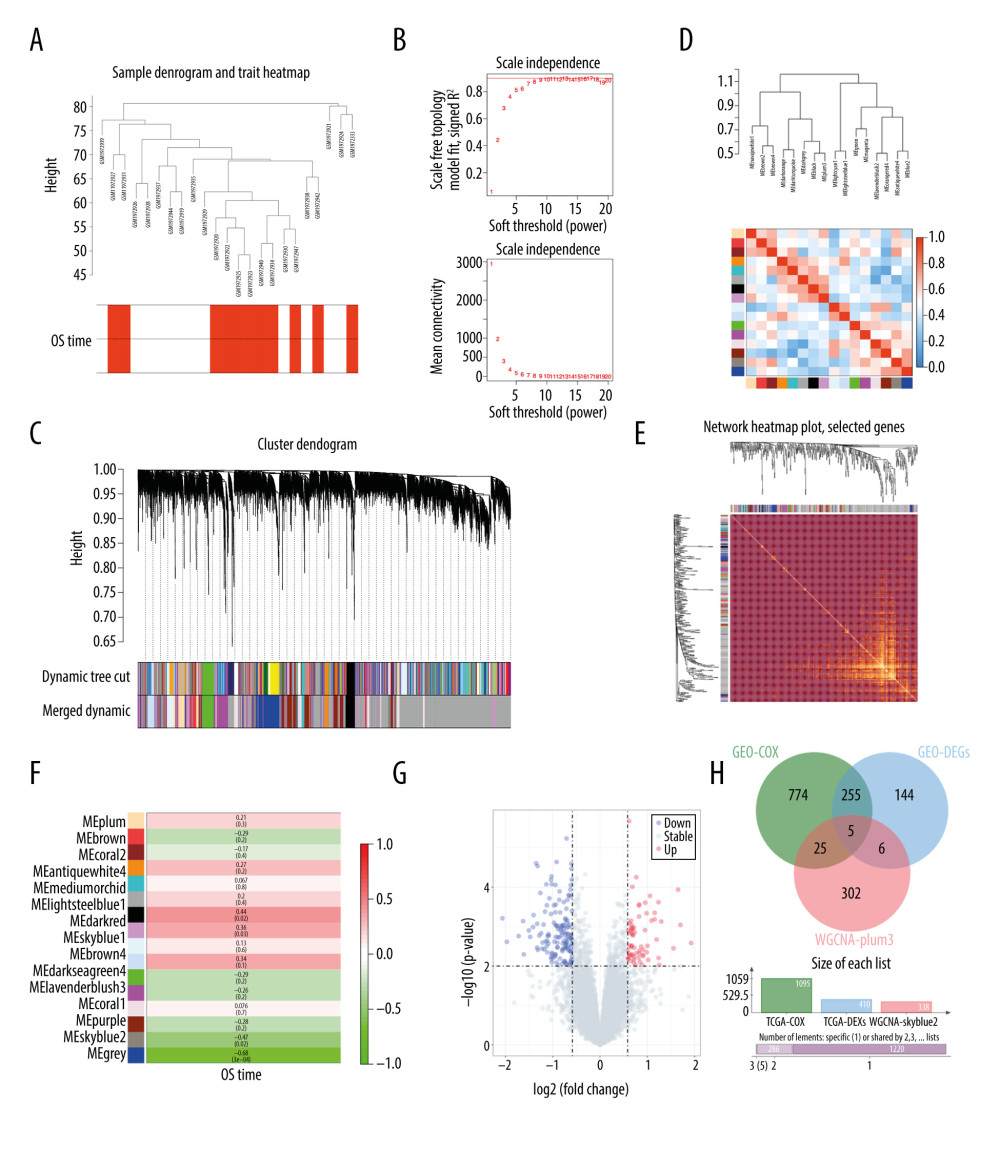 Figure 3. Identification of prognostic genes in the GSE76021 cohort. (A) Cluster dendrogram of samples in the GSE76021 cohort. The dendrogram branches represent the clustered samples. (B) Soft-threshold power analysis of the scale-free fit index (upper) and the mean connectivity (lower). (C) Dendrogram of gene clustering and module assignment. Modules are represented by different colors. (D) Heatmap of adjacencies in the eigengene network. Higher adjacency is represented by red color. (E) Interaction of co-expressing modules. Higher connectivity is represented by light color. (F) Heatmap of the correlation between module eigengenes and patients’ survival. Close relationships are represented by red color. (G) Volcano plot of DEGs between long-term survivors and short-term survivors in the GSE76021 cohort. (H) Venn diagram of the intersection between DEGs, prognostic genes according to Cox analysis, and the WGCNA-plum3 module. WGCNA – weighted correlation network analysis; DEGs – differentially expressed genes.
Figure 3. Identification of prognostic genes in the GSE76021 cohort. (A) Cluster dendrogram of samples in the GSE76021 cohort. The dendrogram branches represent the clustered samples. (B) Soft-threshold power analysis of the scale-free fit index (upper) and the mean connectivity (lower). (C) Dendrogram of gene clustering and module assignment. Modules are represented by different colors. (D) Heatmap of adjacencies in the eigengene network. Higher adjacency is represented by red color. (E) Interaction of co-expressing modules. Higher connectivity is represented by light color. (F) Heatmap of the correlation between module eigengenes and patients’ survival. Close relationships are represented by red color. (G) Volcano plot of DEGs between long-term survivors and short-term survivors in the GSE76021 cohort. (H) Venn diagram of the intersection between DEGs, prognostic genes according to Cox analysis, and the WGCNA-plum3 module. WGCNA – weighted correlation network analysis; DEGs – differentially expressed genes. 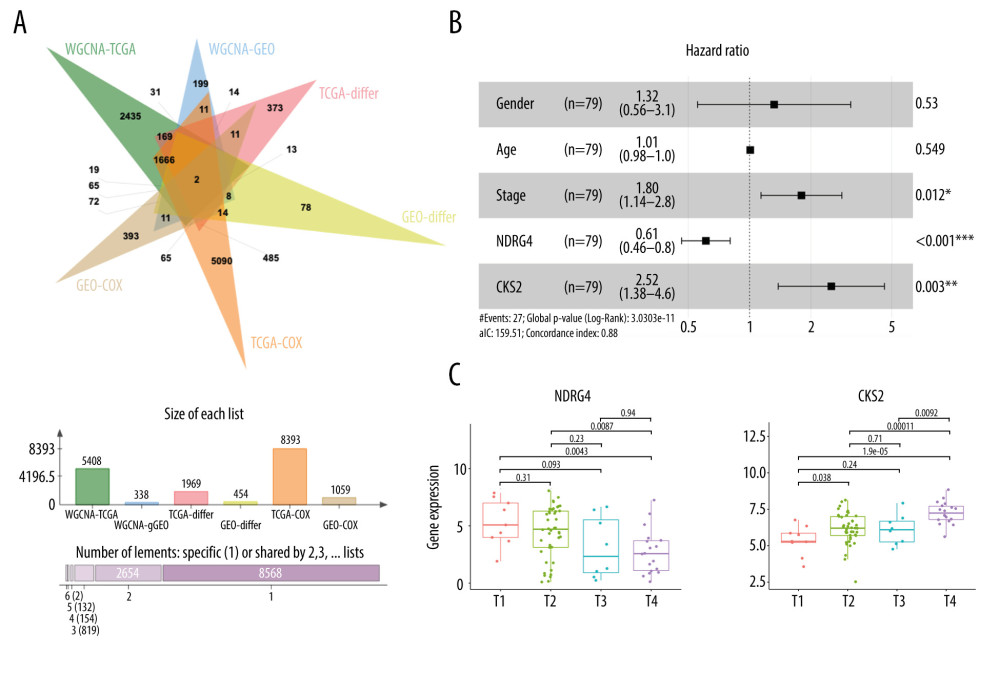 Figure 4. Identification of NDRG4/CKS2 as key prognostic genes in ACC. (A) Venn diagram of the intersection of prognostic genes in the TCGA-ACC cohort and the GSE76021 cohort. (B) Multivariate Cox regression model for overall survival in ACC including gender, age, tumor stage, NDRG4 expression, and CKS2 expression. (C) NDRG4 gene expression in ACC according to different stages. (D) CKS2 gene expression in ACC according to different stages. ACC – adrenocortical carcinoma.
Figure 4. Identification of NDRG4/CKS2 as key prognostic genes in ACC. (A) Venn diagram of the intersection of prognostic genes in the TCGA-ACC cohort and the GSE76021 cohort. (B) Multivariate Cox regression model for overall survival in ACC including gender, age, tumor stage, NDRG4 expression, and CKS2 expression. (C) NDRG4 gene expression in ACC according to different stages. (D) CKS2 gene expression in ACC according to different stages. ACC – adrenocortical carcinoma. 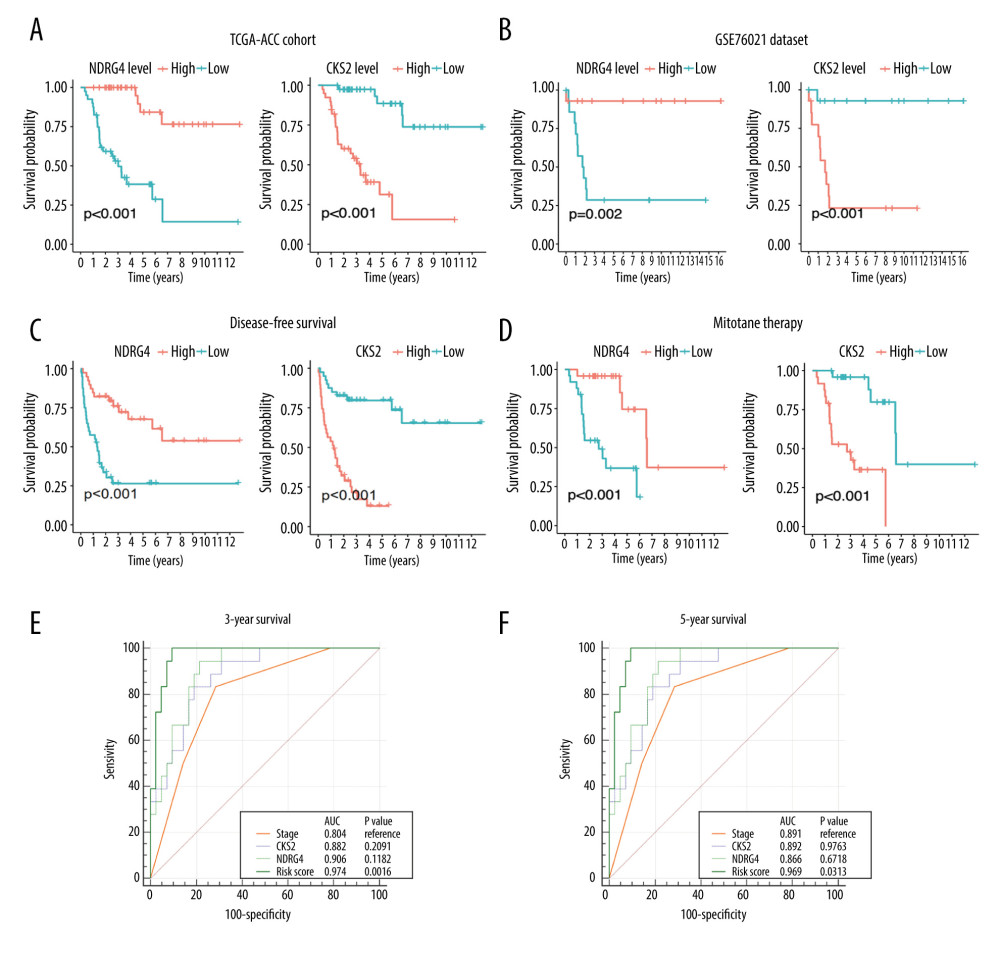 Figure 5. Association between NDRG4/CKS2 expression and survival of ACC patients. (A) Kaplan-Meier survival curve of overall survival in the TCGA-ACC cohort according to NDRG4 and CKS2 expression. (B) Survival curve of overall survival in the GSE76021 cohort according to NDRG4 and CKS2 expression. (C) Survival curve of disease-free survival in the TCGA-ACC cohort according to NDRG4 and CKS2 expression. (D) Survival analysis according to NDRG4 and CKS2 expression in ACC patients treated by mitotane therapy. (E, F) Receiver operating characteristic analysis of the tumor stage, CKS2 expression, NDRG4 expression, and the combined risk score for predicting 3-year survival (E) and 5-year survival (F) in the TCGA-ACC cohort. ACC – adrenocortical carcinoma.
Figure 5. Association between NDRG4/CKS2 expression and survival of ACC patients. (A) Kaplan-Meier survival curve of overall survival in the TCGA-ACC cohort according to NDRG4 and CKS2 expression. (B) Survival curve of overall survival in the GSE76021 cohort according to NDRG4 and CKS2 expression. (C) Survival curve of disease-free survival in the TCGA-ACC cohort according to NDRG4 and CKS2 expression. (D) Survival analysis according to NDRG4 and CKS2 expression in ACC patients treated by mitotane therapy. (E, F) Receiver operating characteristic analysis of the tumor stage, CKS2 expression, NDRG4 expression, and the combined risk score for predicting 3-year survival (E) and 5-year survival (F) in the TCGA-ACC cohort. ACC – adrenocortical carcinoma. 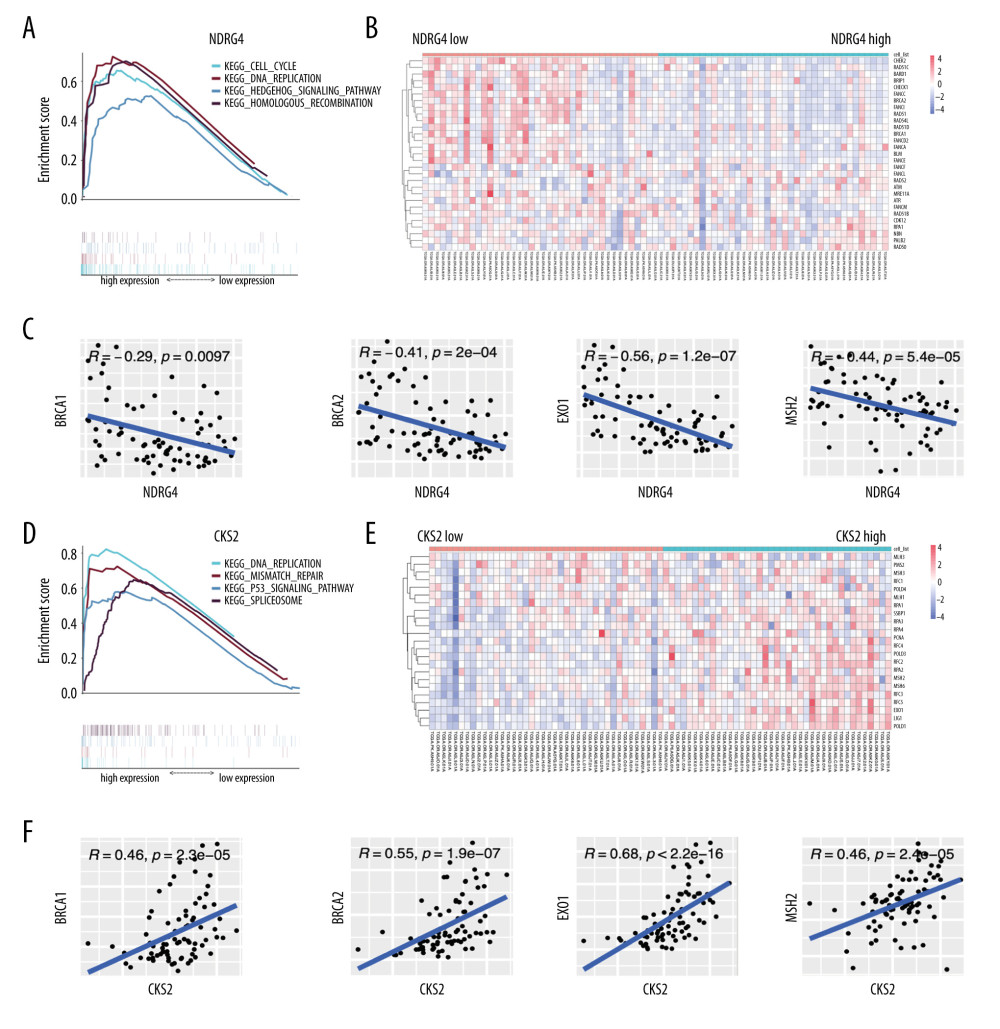 Figure 6. Association between NDRG4/CKS2 expression and homologous recombination/mismatch repair pathways. (A) GSEA analysis of enriched pathways in NDRG4 low-expression samples. (B) Heatmap of genes in the homologous recombination pathway and NDRG4 expression. (C) Association between NDRG4 and BRCA1/BRCA2/EXO1/MSH2 expression in the TCGA-ACC cohort. (D) GSEA analysis of enriched pathways in CKS2 high-expression samples. (E) Heatmap of genes in mismatch repair pathways and CKS2 expression. (F) Association between CKS2 and BRCA1/BRCA2/EXO1/MSH2 expression in the TCGA-ACC cohort. GSEA, gene set enrichment analysis.
Figure 6. Association between NDRG4/CKS2 expression and homologous recombination/mismatch repair pathways. (A) GSEA analysis of enriched pathways in NDRG4 low-expression samples. (B) Heatmap of genes in the homologous recombination pathway and NDRG4 expression. (C) Association between NDRG4 and BRCA1/BRCA2/EXO1/MSH2 expression in the TCGA-ACC cohort. (D) GSEA analysis of enriched pathways in CKS2 high-expression samples. (E) Heatmap of genes in mismatch repair pathways and CKS2 expression. (F) Association between CKS2 and BRCA1/BRCA2/EXO1/MSH2 expression in the TCGA-ACC cohort. GSEA, gene set enrichment analysis. 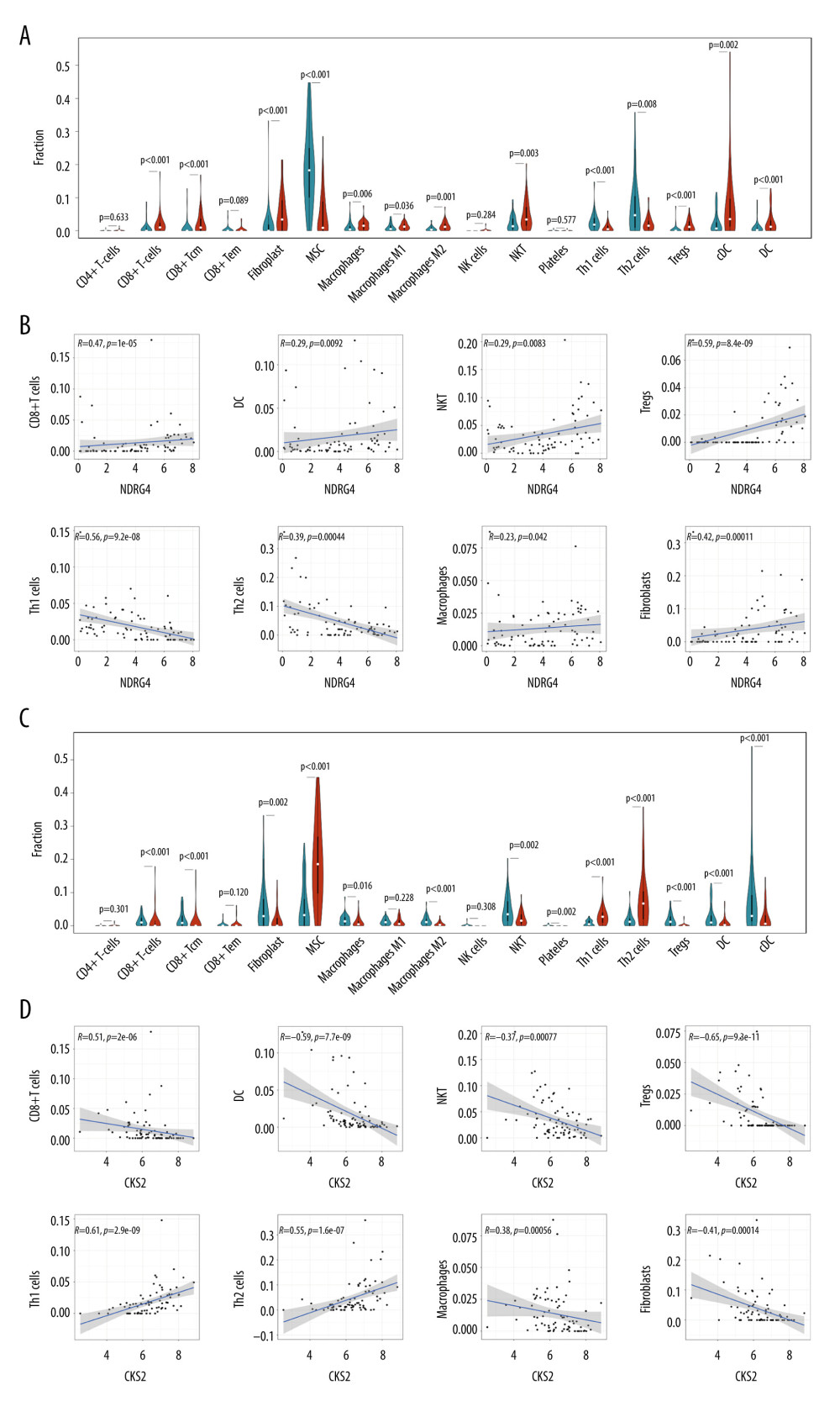 Figure 7. Association between NDRG4/CKS2 expression and immunological status in ACC. (A) Fraction of immune cell infiltration in the NDRG4 high-expression and low-expression samples according to xCell cell type enrichment analysis. (B) Correlation between immune cell infiltration and NDRG4 expression. (C) Fraction of immune cell infiltration in the CKS2 high-expression and low-expression samples according to xCell analysis. (D) Correlation between immune cell infiltration and CKS2 expression. ACC – adrenocortical carcinoma.
Figure 7. Association between NDRG4/CKS2 expression and immunological status in ACC. (A) Fraction of immune cell infiltration in the NDRG4 high-expression and low-expression samples according to xCell cell type enrichment analysis. (B) Correlation between immune cell infiltration and NDRG4 expression. (C) Fraction of immune cell infiltration in the CKS2 high-expression and low-expression samples according to xCell analysis. (D) Correlation between immune cell infiltration and CKS2 expression. ACC – adrenocortical carcinoma. 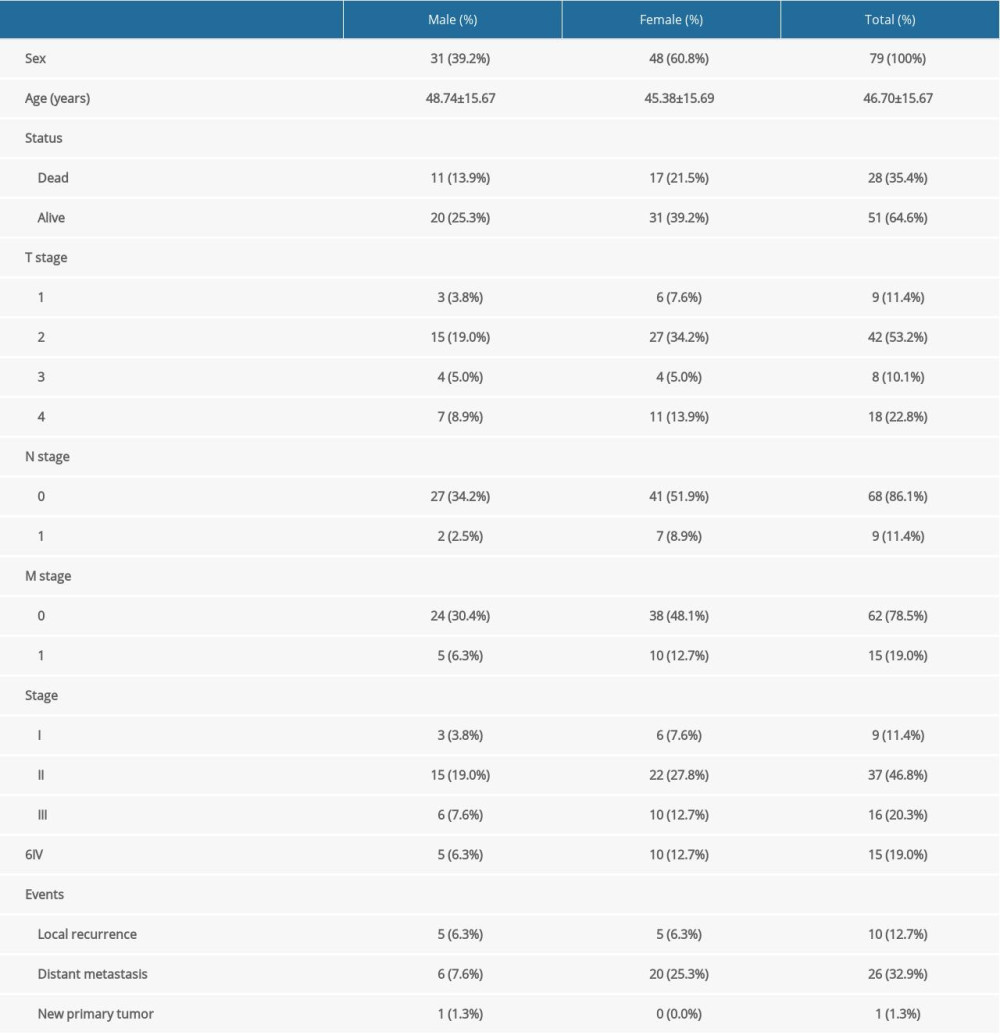
References
1. Crona J, Beuschlein F, Adrenocortical carcinoma – towards genomics guided clinical care: Nat Rev Endocrinol, 2019; 15(9); 548-60
2. Datta J, Roses RE, Surgical management of adrenocortical carcinoma: An evidence-based approach: Surg Oncol Clin N Am, 2016; 25(1); 153-70
3. Else T, Kim AC, Sabolch A, Adrenocortical carcinoma: Endocr Rev, 2014; 35(2); 282-326
4. Erickson LA, Rivera M, Zhang J, Adrenocortical carcinoma: Review and update: Adv Anat Pathol, 2014; 21(3); 151-59
5. Fassnacht M, Johanssen S, Quinkler M, Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: Proposal for a Revised TNM Classification: Cancer, 2009; 115(2); 243-50
6. Margonis GA, Kim Y, Prescott JD, Adrenocortical carcinoma: Impact of surgical margin status on long-term outcomes: Ann Surg Oncol, 2016; 23(1); 134-41
7. Beuschlein F, Weigel J, Saeger W, Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection: J Clin Endocrinol Metab, 2015; 100(3); 841-49
8. Xiao H, Xu D, Chen P, Identification of five genes as a potential biomarker for predicting progress and prognosis in adrenocortical carcinoma: J Cancer, 2018; 9(23); 4484-95
9. Lippert J, Appenzeller S, Liang R, Targeted molecular analysis in adrenocortical carcinomas: A strategy toward improved personalized prognostication: J Clin Endocrinol Metab, 2018; 103(12); 4511-23
10. Sbiera S, Sbiera I, Ruggiero C, Assessment of VAV2 expression refines prognostic prediction in adrenocortical carcinoma: J Clin Endocrinol Metab, 2017; 102(9); 3491-98
11. Subramanian C, Cohen MS, Over expression of DNA damage and cell cycle dependent proteins are associated with poor survival in patients with adrenocortical carcinoma: Surgery, 2019; 165(1); 202-10
12. Pereira SS, Monteiro MP, Bourdeau I, Mechanisms of endocrinology: Cell cycle regulation in adrenocortical carcinoma: Eur J Endocrinol, 2018; 179(2); R95-110
13. Gara SK, Lack J, Zhang L, Metastatic adrenocortical carcinoma displays higher mutation rate and tumor heterogeneity than primary tumors: Nat Commun, 2018; 9(1); 4172
14. Doghman M, Cazareth J, Lalli E, The T cell factor/beta-catenin antagonist PKF115-584 inhibits proliferation of adrenocortical carcinoma cells: J Clin Endocrinol Metab, 2008; 93(8); 3222-25
15. Bernini GP, Moretti A, Viacava P, Apoptosis control and proliferation marker in human normal and neoplastic adrenocortical tissues: Br J Cancer, 2002; 86(10); 1561-65
16. Fay AP, Signoretti S, Callea M, Programmed death ligand-1 expression in adrenocortical carcinoma: An exploratory biomarker study: J Immunother Cancer, 2015; 3; 3
17. Caramuta S, Lee L, Ozata DM, Clinical and functional impact of TARBP2 over-expression in adrenocortical carcinoma: Endocr Relat Cancer, 2013; 20(4); 551-64
18. Zheng S, Cherniack AD, Dewal N, Comprehensive pan-genomic characterization of adrenocortical carcinoma [published correction appears in Cancer Cell, 2016;30(2):363]: Cancer Cell, 2016; 29(5); 723-36
19. Assié G, Letouzé E, Fassnacht M, Integrated genomic characterization of adrenocortical carcinoma: Nat Genet, 2014; 46(6); 607-12
20. Lubitz JA, Freeman L, Okun R, Mitotane use in inoperable adrenal cortical carcinoma: JAMA, 1973; 223(10); 1109-12
21. Terzolo M, Baudin AE, Ardito A, Mitotane levels predict the outcome of patients with adrenocortical carcinoma treated adjuvantly following radical resection: Eur J Endocrinol, 2013; 169(3); 263-70
22. Hermsen IG, Fassnacht M, Terzolo M, Plasma concentrations of o,p’DDD, o,p’DDA, and o,p’DDE as predictors of tumor response to mitotane in adrenocortical carcinoma: Results of a retrospective ENS@T multicenter study: J Clin Endocrinol Metab, 2011; 96(6); 1844-51
23. Sbiera S, Leich E, Liebisch G, Mitotane inhibits Sterol-O-Acyl Transferase 1 triggering lipid-mediated endoplasmic reticulum stress and apoptosis in adrenocortical carcinoma cells: Endocrinology, 2015; 156(11); 3895-908
24. Leek JT, Johnson WE, Parker HS, The sva package for removing batch effects and other unwanted variation in high-throughput experiments: Bioinformatics, 2012; 28(6); 882-83
25. Langfelder P, Horvath S, WGCNA: An R package for weighted correlation network analysis: BMC Bioinformatics, 2008; 9; 559
26. Ritchie ME, Phipson B, Wu D, limma powers differential expression analyses for RNA-sequencing and microarray studies: Nucleic Acids Res, 2015; 43(7); e47
27. Aran D, Hu Z, Butte AJ, xCell: Digitally portraying the tissue cellular heterogeneity landscape: Genome Biol, 2017; 18(1); 220
28. Zhou RH, Kokame K, Tsukamoto Y, Characterization of the human NDRG gene family: A newly identified member, NDRG4, is specifically expressed in brain and heart: Genomics, 2001; 73(1); 86-97
29. Schilling SH, Hjelmeland AB, Radiloff DR, NDRG4 is required for cell cycle progression and survival in glioblastoma cells: J Biol Chem, 2009; 284(37); 25160-69
30. Yang X, An L, Li X, NDRG3 and NDRG4, two novel tumor-related genes: Biomed Pharmacother, 2013; 67(7); 681-84
31. Ding W, Zhang J, Yoon JG, NDRG4 is downregulated in glioblastoma and inhibits cell proliferation: OMICS, 2012; 16(5); 263-67
32. Xiao W, Zhao H, Dong W, Quantitative detection of methylated NDRG4 gene as a candidate biomarker for diagnosis of colorectal cancer: Oncol Lett, 2015; 9(3); 1383-87
33. Chu D, Zhang Z, Zhou Y, NDRG4, a novel candidate tumor suppressor, is a predictor of overall survival of colorectal cancer patients: Oncotarget, 2015; 6(10); 7584-96
34. Kotipatruni RP, Ren X, Thotala D, Jaboin JJ, NDRG4 is a novel oncogenic protein and p53 associated regulator of apoptosis in malignant meningioma cells: Oncotarget, 2015; 6(19); 17594-604
35. Agosta C, Laugier J, Guyon L, MiR-483-5p and miR-139-5p promote aggressiveness by targeting N-myc downstream-regulated gene family members in adrenocortical cancer: Int J Cancer, 2018; 143(4); 944-57
36. Spruck CH, de Miguel MP, Smith AP, Requirement of Cks2 for the first metaphase/anaphase transition of mammalian meiosis: Science, 2003; 300(5619); 647-50
37. Kang MA, Kim JT, Kim JH, Upregulation of the cycline kinase subunit CKS2 increases cell proliferation rate in gastric cancer: J Cancer Res Clin Oncol, 2009; 135(6); 761-69
38. Lan Y, Zhang Y, Wang J, Aberrant expression of Cks1 and Cks2 contributes to prostate tumorigenesis by promoting proliferation and inhibiting programmed cell death: Int J Cancer, 2008; 123(3); 543-51
39. Chen R, Feng C, Xu Y, Cyclin-dependent kinase-associated protein Cks2 is associated with bladder cancer progression: J Int Med Res, 2011; 39(2); 533-40
40. Liberal V, Martinsson-Ahlzén HS, Liberal J, Cyclin-dependent kinase subunit (Cks) 1 or Cks2 overexpression overrides the DNA damage response barrier triggered by activated oncoproteins: Proc Natl Acad Sci USA, 2012; 109(8); 2754-59
41. Chehade M, Bullock M, Glover A, Key MicroRNA’s and their targetome in adrenocortical cancer: Cancers (Basel), 2020; 12(8); 2198
Errate

Figures
Figure 1. Flow chart of the study design.Figure 2. Identification of prognostic genes in the TCGA-ACC cohort by WGCNA. (A) Cluster dendrogram of samples in the TCGA-ACC cohort to detect outliers. The dendrogram branches represent the clustered samples. (B) Soft-threshold power analysis of the scale-free fit index (upper) and the mean connectivity (lower). (C) Dendrogram of gene clustering and module assignment. Modules are represented by different colors. (D) Heatmap of adjacencies in the eigengene network. Higher adjacency is represented by red color. (E) Interaction of co-expressing modules. Higher connectivity is represented by light color. (F) Heatmap of the correlation between module eigengenes and patients’ survival. Close relationships are represented by red color. (G) Volcano plot of DEGs between long-term survivors and short-term survivors in the TCGA-ACC cohort. (H) Venn diagram of the intersection of DEGs between long-term survivors and short-term survivors, prognostic genes according to univariate Cox regression model, and the WGCNA-skyblue2 module. WGCNA – weighted correlation network analysis; DEGs – differentially expressed genes.Figure 3. Identification of prognostic genes in the GSE76021 cohort. (A) Cluster dendrogram of samples in the GSE76021 cohort. The dendrogram branches represent the clustered samples. (B) Soft-threshold power analysis of the scale-free fit index (upper) and the mean connectivity (lower). (C) Dendrogram of gene clustering and module assignment. Modules are represented by different colors. (D) Heatmap of adjacencies in the eigengene network. Higher adjacency is represented by red color. (E) Interaction of co-expressing modules. Higher connectivity is represented by light color. (F) Heatmap of the correlation between module eigengenes and patients’ survival. Close relationships are represented by red color. (G) Volcano plot of DEGs between long-term survivors and short-term survivors in the GSE76021 cohort. (H) Venn diagram of the intersection between DEGs, prognostic genes according to Cox analysis, and the WGCNA-plum3 module. WGCNA – weighted correlation network analysis; DEGs – differentially expressed genes.Figure 4. Identification of NDRG4/CKS2 as key prognostic genes in ACC. (A) Venn diagram of the intersection of prognostic genes in the TCGA-ACC cohort and the GSE76021 cohort. (B) Multivariate Cox regression model for overall survival in ACC including gender, age, tumor stage, NDRG4 expression, and CKS2 expression. (C) NDRG4 gene expression in ACC according to different stages. (D) CKS2 gene expression in ACC according to different stages. ACC – adrenocortical carcinoma.Figure 5. Association between NDRG4/CKS2 expression and survival of ACC patients. (A) Kaplan-Meier survival curve of overall survival in the TCGA-ACC cohort according to NDRG4 and CKS2 expression. (B) Survival curve of overall survival in the GSE76021 cohort according to NDRG4 and CKS2 expression. (C) Survival curve of disease-free survival in the TCGA-ACC cohort according to NDRG4 and CKS2 expression. (D) Survival analysis according to NDRG4 and CKS2 expression in ACC patients treated by mitotane therapy. (E, F) Receiver operating characteristic analysis of the tumor stage, CKS2 expression, NDRG4 expression, and the combined risk score for predicting 3-year survival (E) and 5-year survival (F) in the TCGA-ACC cohort. ACC – adrenocortical carcinoma.Figure 6. Association between NDRG4/CKS2 expression and homologous recombination/mismatch repair pathways. (A) GSEA analysis of enriched pathways in NDRG4 low-expression samples. (B) Heatmap of genes in the homologous recombination pathway and NDRG4 expression. (C) Association between NDRG4 and BRCA1/BRCA2/EXO1/MSH2 expression in the TCGA-ACC cohort. (D) GSEA analysis of enriched pathways in CKS2 high-expression samples. (E) Heatmap of genes in mismatch repair pathways and CKS2 expression. (F) Association between CKS2 and BRCA1/BRCA2/EXO1/MSH2 expression in the TCGA-ACC cohort. GSEA, gene set enrichment analysis.Figure 7. Association between NDRG4/CKS2 expression and immunological status in ACC. (A) Fraction of immune cell infiltration in the NDRG4 high-expression and low-expression samples according to xCell cell type enrichment analysis. (B) Correlation between immune cell infiltration and NDRG4 expression. (C) Fraction of immune cell infiltration in the CKS2 high-expression and low-expression samples according to xCell analysis. (D) Correlation between immune cell infiltration and CKS2 expression. ACC – adrenocortical carcinoma. In Press
15 Apr 2024 : Laboratory Research
The Role of Copper-Induced M2 Macrophage Polarization in Protecting Cartilage Matrix in OsteoarthritisMed Sci Monit In Press; DOI: 10.12659/MSM.943738
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
18 Apr 2024 : Clinical Research
Comparative Analysis of Open and Closed Sphincterotomy for the Treatment of Chronic Anal Fissure: Safety an...Med Sci Monit In Press; DOI: 10.12659/MSM.944127
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952