05 December 2021: Database Analysis
Alternative Splicing Events in Immune Infiltration of Lung Adenocarcinoma
Tianpeng HuangDOI: 10.12659/MSM.934491
Med Sci Monit 2021; 27:e934491






Abstract
BACKGROUND: The exact mechanisms of lung adenocarcinoma (LUAD) etiology and pathogenesis remain unclear.
MATERIAL AND METHODS: In this study, we evaluated alternative splicing (AS) events in LUAD. We separately analyzed LUAD data from The Cancer Genome Atlas (TCGA) database and AS-related feature lists from SpliceSeq to develop risk models for AS events and further validated risk models for AS events. The association between immune-related features and the risk model of AS events was evaluated.
RESULTS: We found that exon skip (ES) and mutually exclusive exons (ME) exhibited the most and least AS events, respectively. The risk score and stage of LUAD patients were strongly associated with prognosis and were independent influences on the prognosis of LUAD. The pathological stage (stage, T, N) and risk model were incorporated to construct a column line graph with better predictive ability. Risk models were associated with tumor microenvironment, and higher risk score values were associated with higher M2 macrophage content and lower M0 macrophage and helper T lymphocyte content. The correlation between core genes and immunity was further assessed by analyzing differentially-expressed genes between risk models. These results suggest an association between the level of risk for AS events and the density of immune cell infiltration.
CONCLUSIONS: Our findings suggest a clear association between AS risk model and patient prognosis, and was performed to confirm the biological relationship between AS and immunity.
Keywords: Adenocarcinoma of Lung, Alternative Splicing, Anesthesia, Local, Immunotherapy, Female, Humans, Lung, Lung Neoplasms, Male
Background
Lung cancer (LC) has the highest incidence and mortality rates among malignant tumors worldwide, with lung adenocarcinoma (LUAD), the predominant pathological type of LC, causing a great burden of disease [1]. Due to the lack of sensitivity of diagnostic tools and the limitations of diagnostic conditions, 75% of patients are at advanced stages at the time of diagnosis [2]. Prognostic assessment of LUAD and individualized treatment are very important to reduce metastasis and recurrence and to prolong survival [2]. It is gradually being accepted that LUAD recognition is not a sudden change, but the result of a combination of genetic and epigenetic changes playing a role [3]. Therefore, there is an urgent need to understand the molecular mechanisms of LUAD. With the advent of next-generation sequencing technologies, genes involved in the onset, progression, and metastasis of LUAD, such as KRAS, EGFR, ALK, and ROS1, are currently an important topic of research that can improve early screening and more precise individualized treatment of LUAD [1,3].
Recent studies have gradually established that alternative splicing (AS) is also responsible for carcinogenesis [4]. LUAD tissues exhibit more aberrant activation patterns of AS; however, the exact role of AS in lung cancer is unknown [5]. In recent years, epigenetic and transcriptomic studies have identified many valuable tumor markers that may provide new ideas for future diagnosis and treatment [6–8]. Immune response affects tumor development and sensitivity to drugs, while AS influences the type and function of immune cells [9,10]. In this study, we established an AS-related risk model by use of the bioinformatics method to further analyze the correlation between the risk model and immunity, and provide new ideas for future in-depth research. The workflow of this study is shown in Figure 1.
Material and Methods
DATA ACQUISITION:
The expression data and corresponding clinical information of LUAD were downloaded from the TCGA database (
AS events can be divided into 7 basic types: alternate acceptor site AA), alternate donor site (AD), alternate promoter (AP), alternate terminator (AT), exon skipping (ES), exon mutual exclusion (ME), and retained intron (RI) [11]. Seven types of AS events of LUAD were downloaded from the TCGA SpliceSeq database (https://bioinformatics.mdanderson.org/TCGASpliceSeq/). Each AS event was quantified by the percentage splice (PSI) value, which ranged from 0 to 1 [11]. AS events that met the following criteria were included for the next step of analysis, with PSIvalue ≥75%. The visualization of interaction of AS events was performed through UpSet plots using the UpSet package.
Further, the AS dataset was processed with survival information (survival status and overall survival) by removing blank samples and merging. To identify AS events associated with survival in LUAD, univariate Cox proportional risk regression analysis was performed.
AS-RELATED PROGNOSTIC RISK MODEL:
The prognosis-related AS genes identified above were further analyzed.
Minimum absolute shrinkage and selection operator (LASSO) and multivariate Cox proportional risk regression analysis were performed using the glmnet package and survival package to narrow down the candidate genes, calculate the coefficient, and build a prognostic risk model. The penalty parameter (λ) was determined by the minimum parameter. The risk score was calculated using the following equation:
Coefi: correlation coefficient of the identified AS events, PSIi: PSI of the AS identified events.
The risk score in each sample was calculated and divided into a high-risk score subgroup (>median value) and a low-risk score subgroup (<median value). The results were constructed by visualizing distribution curves for risk score subgroup, survival distributions, and model gene expression heatmap. Kaplan-Meier analysis was used to construct survival curves using the survival package and SurvMiner package between 2 subgroups, and the log-rank test was performed.
ASSOCIATION OF THE RISK SCORE WITH CLINICAL CHARACTERISTICS:
To determine the relationship with prognosis in LUAD, univariate Cox proportional risk regression analysis was performed for several factors, including age, sex, clinical stage (TMN), and risk score. To exclude the interference of confounding factors, multivariate Cox proportional risk regression analysis was performed for the above-mentioned multiple influencing factors. The above process was performed in the survival package, and
The Limma package was used to analyze the differences between the risk score subgroups and several factors, including age, sex, and clinical stage (TMN). The Wilcoxon rank sum test was used to compare the 2 subgroups, and the Kruskal-Wallis test was used to compare 3 or more subgroups.
A nomogram can be used as a multiple-indicator combination to diagnose or predict disease onset or progression [12]. The rms package was used to plot the column line graph constructs, and then the mean of the predicted survival probability and the corresponding actual survival probability (calculated by the Kaplan-Meier method) were calculated separately for each group of study subjects, and the 2 were combined to plot the 4 calibration points, and finally the 4 calibration points were connected to obtain the prediction calibration curve. If the prediction calibration curve is closer to the standard curve, the better predictive ability of the column line plot is indicated.
CORRELATION OF RISK SCORE SUBGROUP WITH IMMUNITY:
The ESTIMATE package was used to assess tumor microenvironment (TME) while generating 4 measures (stromal score, immune score, estimate score, and tumor purity). The higher the score of immune cells or stromal cells, the more immune or stromal components in TME. The Wilcoxon rank sum test was used to compare the differences in stromal score, immune score, estimate score, and tumor purity between the high-risk score group and low-risk score group using the Limma package, and
The CIBERSORT package was used to obtain the content of immune infiltrating cells in each LUAD sample, including 22 immune cells such as CD8T cells, activated dendritic cells, macrophages, natural killer T cells, and regulatory T cells. The Wilcoxon rank test was used to compare the difference between risk score subgroup and immune infiltrating cells. The correlation between risk score and significantly different immune cells was further compared by Spearman’s method.
Single-gene enrichment analysis (ssGSEA) was used to quantify the relative abundance of immune infiltrating cells and immune-related functions in each sample using the immune score. The samples were first analyzed by ssGSEA using the GSVA package to obtain an immune score in the LUAD dataset. Then, the Wilcoxon rank test was used to compare the differences in immune score between the risk score subgroups using the Limma package.
Immune check checkpoints were included, such as BTNL2, LAIR1, CD44, IDO2, HHLA2, TNFRSF14, TNFRSF25, and TNFSF18. Differences in immune checkpoints between the high-risk score subgroup and low-risk score subgroup were compared by Limma package using Wilcoxon rank test, and
DIFFERENTIAL GENES BETWEEN RISK SCORE SUBGROUP:
The Limma package was used to apply differential analysis to identify differentially-expressed genes (DEGs) between high- and low-risk score groups, and DEGs with statistically significant differences were screened by logFC >1 and
Kaplan-Meier method for predictive analysis between groups, and the log-rank test was used to determine the significance of differences.
CORRELATION BETWEEN DIFFERENTIALLY-EXPRESSED GENES AND IMMUNITY:
The Wilcoxon test was applied using the Limma package to compare the association between DEGs and immunity, including immune checkpoint, tumor microenvironment, 22 immune infiltrating cells, and immune score. The preparation materials for the above process were the same as the previous steps, and
ALTERNATIVE SPLICING-ALTERNATIVE SPLICING FACTORS NETWORK:
The expression of alternative slicing factors in transcriptional sampling data was extracted and processed uniformly. The correlation of survival-related AS and alternative splicing factors were analyzed using the Spearman method with
STATISTICAL ANALYSIS:
All data were processed using R software (version 4.0.3). Data extraction was performed using PERL (version 5.8.3). Network plots were created using Cytoscape (version 3.7.2).
Results
IDENTIFICATION OF AS-RELATED EVENTS OF LUAD:
Seven AS events types were included in this study: AA, AD, AP, AT, ES, ME, and RI. A total of 513 cohorts of LUAD tissues and 59 cohorts of paraneoplastic tissues from the TCGA database were included in this study (Supplementary Table 1). A total of 43 948 AS events were detected, including 16 793 ES events in 1744 genes, 8992 AP events in 732 genes, 8546 AT events in 1099 genes, 3559 AA events in 224 genes, 3057 AD events in 224 genes, and 2781 RI events in 298 genes. The UpSet plot shows the interaction of genes with different AS events, with ES being the most common AS events (Figure 2A). A significant proportion of these genes contained more than 2 AS events, and up to 7 different AS events occurred in a single gene. To identify the relationship between AS and survival, further univariate Cox proportional risk regression analysis was performed (Supplementary Table 2). We found that 19 45 AS events and the corresponding 1408 genes were significantly associated with the overall survival of LUAD patients (Figure 2B). According to the priority ranking of AS events, ES events and AP events were the prognosis-related AS events that occupied a larger proportion. Most genes have only 1 AS event, and a few genes have more than 2 different splicing patterns. Volcano plots were used to identify AS prognosis-related genes in LUAD (Figure 2C), and the top 20 AS events of these 7 types of events are demonstrated by bubble plots in Supplementary Figure 1.
CONSTRUCTION OF RISK MODEL:
The prognosis-related AS events were reduced by LASSO regression analysis (Supplementary Figure 2). Further, multivariate Cox regression analysis was performed to obtain a list of prognostic characteristics of AS events, and risk scores were calculated according to the weight of each event (Supplementary Table 3). As shown in Figure 3A, the number of patient deaths increased with higher risk score values. The heatmap in Figure 3B demonstrates the differential expression of identified AS events in the risk score subgroups. The identified AS events were unevenly distributed between high- and low-risk subgroups. Kaplan-Meier survival curve analysis showed that the high-risk score group was associated with poorer OS (P<0.001, Figure 3C). The ROC curve analyzed the relationship between risk model and survival at 1, 2, and 3 years, which were 0.771, 0.770, and 0.784, respectively, indicating that the risk prognostic model had high predictive ability (Figure 3D).
ASSOCIATION OF RISK MODEL WITH CLINICAL CHARACTERISTICS:
To better assess the correlation between the risk score and clinical characteristics, the independent variables were included in the Cox regression analysis, and the continuous variable was risk score, and the categorical variables were sex, age, and clinical stage (Stage). Then, univariate and multivariate Cox regression analyses were performed for the above variables. The results of the univariate Cox proportional risk regression analysis showed that risk score and clinical stage (Stage) were the risk factors affecting overall cancer survival (P<0.001, Figure 4A). The results of multivariate Cox regression model analysis showed that risk score and clinical stage (Stage) were independent factors influencing the prognosis of LUAD patients (P<0.05, Figure 4B). In conclusion, the risk score showed strong potential for clinical application.
Further analysis of differences in risk scores and clinical characteristics (age, sex, and clinical stage) was performed, and the risk score was significantly associated with sex, clinical stage (T), clinical stage (N), and clinical stage (Stage) (P<0.05, Figure 4C). The variables clinical stage (T), clinical stage (N), clinical stage (Stage), and risk score were included to construct a risk prediction model using column line plots (Figure 4D). A calibration chart shows that the closer the prediction calibration curve is to the standard curve, the better the predictive ability of the column line plot is.
ASSOCIATION OF RISK MODEL WITH IMMUNITY:
The ESTIMATE package was used to calculate the immune score, stromal score, estimate score, and tumor purity. Compared to the high-risk score group, the low-risk score group had more stromal and immune components in TME and lower tumor purity (P <0.05, Figure 5A).
The CIBERSPOT package was used to assess the contents of 22 immune infiltrating cells and further compare the relationship between the risk model and immune infiltrating cells, showing significant differences between the risk score subgroup in terms of M0 macrophages, M2 macrophages, and helper T cells (P<0.05, Figure 5B). Further correlation analysis showed that higher risk score values were associated with higher M2 macrophage content and with lower M0 macrophage and helper T lymphocyte content (P<0.05, Supplementary Figure 3).
Calculating immune scoring showed the risk score subgroups were significantly different in dendritic cells, B lymphocytes, major histocompatibility complex class I, type II interferon response, tumor-infiltrating lymphocyte, helper T lymphocytes, regulatory T cells, human leukocyte antigen, mast cells, neutrophils, and natural killer cells (P<0.05, Figure 5C).
BTNL2, LAIR1, CD44, IDO2, HHLA2, TNFRSF14, TNFRSF25, TNFSF18, CD80, ICOS, ADORA2A, TNFSF15, CTLA4, CD27, CD200R1 CD200, LAG3, BTLA, CD40LG, NRP1, CD48, CD28, CD160 were more expressed in the low-risk scoring group, while CD276 was more expressed in the high-risk scoring group (P<0.05, Figure 5D).
DIFFERENTIAL GENE ANALYSIS:
The expression of TTC39C, CDKN2A, and PKIB in paraneoplastic tissues was significantly lower than that in LUAD tissues, and the difference was statistically significant (P<0.001, Figure 6A). Patients with highly expressed PKIB and CDKN2A had shorter survival, while those with high expression of TTC39C had longer survival (P<0.001, Figure 6A). Further, PKIB was significantly different between N0 and N1 (P<0.05, Figure 6B), CDKN2A did not show significant differences in sex, age or clinical stage (Figure 6C), and TTC29C was significantly different in sex, T2, and T3 (P<0.05, Figure 6D).
COMPREHENSIVE ANALYSIS OF DEGS AND IMMUNITY:
First, the relationship between DEGs and tumor microenvironment was assessed. Compared with the low-expression group, the immune cell score and stromal cell score were higher in the high-expression PKIB group (P<0.001, Figure 7A). CDKN2A was not significantly correlated with immune cell score and stromal score (Figure 7B). Compared with the high-expression group, the immune cell score and stromal score were higher in the low-expression TTC29C group (P<0.001, Figure 7C).
Subsequently, further analysis of the association between CDKN2A, PKIB, TTC29C, and immune scoring also revealed that the expression of PKIB and TTC29C are significantly correlated with most multiple types of immune cells and immune function (P<0.05, Figure 8A). Then, we assessed the relationship between DEGs and 22 immune infiltrating cells. The expression of PKIB was differential in resting state mast cells, M2 macrophages, mast cells, activated NK cells, and B lymphocytes; TTC39C was differentially expressed in plasma cells, resting mast cells; CDKN2A was positively correlated with activated CD4+ T memory T cells and M1 macrophages (P<0.05, Figure 8B). In addition, PKIB, CDKN2A, and TTC29C were correlated with most immune checkpoints (P<0.05, Figure 8C). Therefore, PKIB, CDKN2A, and TTC29C may become immunotherapeutic targets.
CONSTRUCTION OF ALTERNATIVE SPLICING-ALTERNATIVE SPLICING FACTORS NETWORK:
A number of alternative splicing factors were included, such as ACIN1, AGGF1, ALYREF, AQR, ARGLU1, BAG2, BCAS1, and BCAS2. By performing correlation analysis between AS and alternative splicing factors, we found that alternative splicing factors DDX39B, SEC31B, and CLK1, and a series of AS exist in a network of interactions that together play a role in tumor development (Figure 9).
Discussion
In this study, an AS-related risk model was constructed using a series of prognosis-related AS signatures. First, survival-related AS signatures were screened by the LASSO and Cox regression model constructs, which were more stable and reliable compared with a single method. The AUCs of 0.771, 0.770, and 0.784 at 1, 2, and 3 years, respectively, were confirmed by internal tests, demonstrating the strong predictive ability of the AS-related risk model. Further, risk score and clinical staging (Stage) were found to be significantly associated as independent influences on the prognosis of LUAD patients. Clinical staging (TNM) is widely used to assess the prognosis of LUAD patients, but lacks individuality. Therefore, the construction of columnar maps to predict the prognosis of LUAD patients based on the patient’s profile can be helpful for clinical prediction. Unfortunately, columnar maps have been shown to have good predictive power for LUAD, but due to their limitations, they cannot accurately project survival in LUAD and still need further refinement.
The process of tumor formation is often accompanied by the formation of a tumor bed, changes in the surrounding connective tissue and stromal cells, and, ultimately, a microenvironment suitable for cancer cell growth [13]. Key components of the stroma in the tumor microenvironment play an important role in several pathological processes, including cancer cell growth and metastasis, and influence the body’s anti-tumor immune response [14]. In the tumor microenvironment, immune cells instead serve as a protective umbrella for tumor cells by interfering with the body’s immune system. In addition, immune editing by cancer cells can lead to “immune escape” [15]. Studies have shown that lymphocytes, macrophages, dendritic cells, and NK cells are associated with the prognosis of LUAD and play important roles in anti-tumor, immune escape, and promotion of tumor growth and migration [16–19]. The present study found that the AS-related risk model reflected the tumor microenvironment, which may provide new ideas about LUAD pathological changes.
Immune infiltrating cells are associated with tumor growth, infiltration, and invasion [20]. Using the CIBERSPOT package to demonstrate the content of 22 immune infiltrating cells, it was found that the content of immune infiltrating cells was different in the AS risk score subgroups, with the low-risk score group having more M0 macrophages and helper T lymphocytes and fewer M2 macrophages. It was further found that the low-risk score subgroup was more associated with prognosis than was the high-risk score subgroup. The relevance of macrophage infiltration in the tumor microenvironment has long been known. According to the “macrophage homeostasis theory”, there are be 2 types of macrophages with different functions in the tumor microenvironment: M1 type, the classically activated macrophages, and M2 type, the alternative activated macrophages. The M1 type is involved in antigen delivery and tumor cell killing by secreting various antigenic molecules and inflammatory factors and expressing MHC II and B7 molecules, while the M2 type can have the effect of promoting the pathological process of cancer cells. Helper T lymphocytes can fight against tumors by secreting cytokines (eg, IL-2, IFN-r, and TBF-B), killing tumor cells independently of CTL, and expressing CD40 ligands to stimulate CTL by binding to antigen-presenting cells CD40 [18]. Differences in immune infiltrating cells in high- and low-risk score groups are associated with the prognosis of LUAD patients, further suggesting that AS may influence the composition of the tumor microenvironment.
Immunotherapy is one of the more successful approaches in cancer treatment in recent years. In this study, the low-risk score subgroup had more immune checkpoints and a better response to immunotherapy compared to the high-risk score subgroup. The risk model possessed a good predictive power for immunotherapy. In addition, a noteworthy point is that CD276 was more highly expressed in the high-risk score subgroup. CD276 is overexpressed in tumors, including melanoma, leukemia, breast, prostate, and colorectal cancers, which is associated with poor prognosis and worse clinical outcome, and is a promising target for tumor immunotherapy and a new hope for immunotherapy-insensitive LUAD [21].
The core genes in the risk model were further screened by differential analysis. The tetratricopeptide repeat domain 39C (TTC39C) is associated with diabetic maculopathy with vision loss and skeletal muscle atrophy [22]. The mechanism by which TTC39C plays a role in cancer is unclear, but there is an association. Overexpression of TTC39C can lead to impaired ERK1/2 MAP kinase and MAP transduction signaling [23]. The relationship between the MAPK signaling pathway and a variety of tumors is becoming known with the discovery of multiple proteins in the MAP transduction signaling pathway. cAMP-dependent protein kinase inhibitor-B (PKIB), a protein kinase inhibitor, is a class of proteins that inhibit cAMP-dependent protein kinase activity [24–26]. One investigator found that PKIB expression is upregulated in NSCLC and acts as an important effector of NSCLC cell proliferation and metastasis through the PI3K/Akt pathway [27]. The cyclin-dependent kinase inhibitor 2A (CDKN2A), located on chromosome 9p21, encodes the p16 protein. p16 acts as a cell cycle regulator that inhibits cell cycle proteins, thereby preventing premature transition from G1 to S phase [28]. CDKN2A is associated with familial atypical melanoma multiforme syndrome and familial pancreatic cancer [28]. Further internal datasets validated that TTC39C, CDKN2A, and PKIB are associated with survival and may be prognostically relevant therapeutic targets for LUAD. In addition, the core genes and immune presence associations among risk models were assessed, showing that AS plays an important role in the tumor microenvironment.
Song et al used LASSO-Cox regression analysis to construct a multiple alternative splicing prognostic risk model of LUAD and a single alternative splicing prognostic risk model of LUAD, respectively, and found that the multiple alternative splicing prognostic risk models prognostic risk models possessed better predictive ability [29]. The risk model constructed for multiple alternative splicing events alone in this study also yielded satisfactory results. In addition, multiple quantities including risk scores were incorporated to construct and validate the reliability of column line graphs. Li et al constructed the alternative splicing risk prognostic model of LUAD by Cox risk regression analysis and also obtained more satisfactory results [30]. Different methods exist for constructing risk prognostic models, and the reliability of the risk prognostic models of alternative splicing-related LUAD constructed under different methods can be compared in the future. There are still shortcomings in this study, and the constructed risk model needs a larger sample to confirm the predictive ability in clinical practice. There is little research on the mechanism of action of the identified core genes in LUAD, and further studies are needed to confirm the role of core genes.
Conclusions
In this study, a dataset from public databases was used to build and evaluate AS risk models and further elucidate the relationship with prognosis of LUAD patients. In addition, an in-depth mechanistic analysis was performed to confirm the biological relationship between AS and immunity.
Figures
 Figure 1. Flow chart of our study. Visio version 1.5.6.
Figure 1. Flow chart of our study. Visio version 1.5.6. 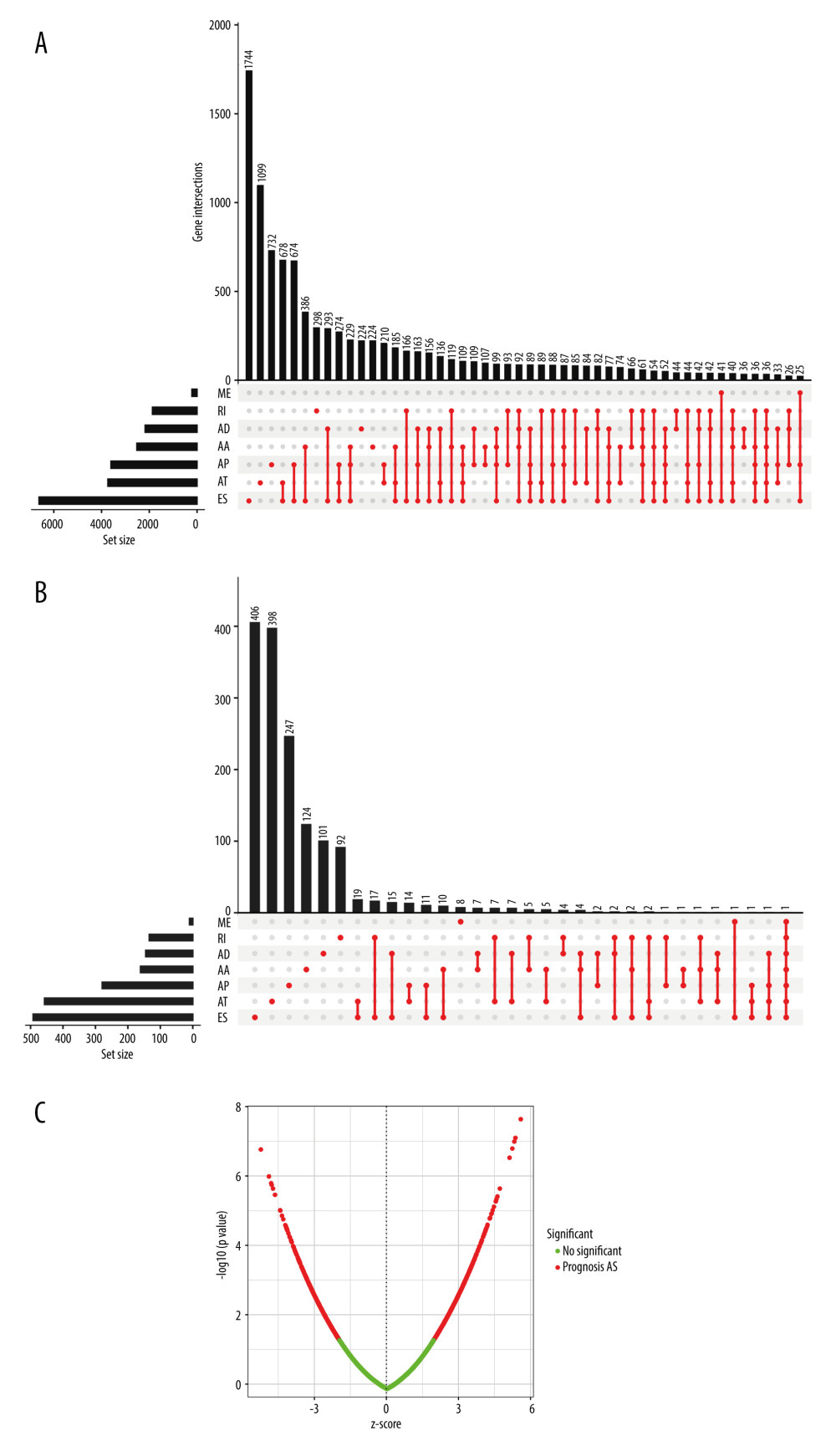 Figure 2. Alternative splicing (AS) of lung adenocarcinoma (LUAD) analysis. (A) UpSet demonstrates the interaction of AS events. (B) UpSet demonstrates the interaction of Prognosis-related AS events. (C) Volcano plot to show prognosis-related AS events. R Software version 4.0.3 (https://www.r-project.org/).
Figure 2. Alternative splicing (AS) of lung adenocarcinoma (LUAD) analysis. (A) UpSet demonstrates the interaction of AS events. (B) UpSet demonstrates the interaction of Prognosis-related AS events. (C) Volcano plot to show prognosis-related AS events. R Software version 4.0.3 (https://www.r-project.org/). 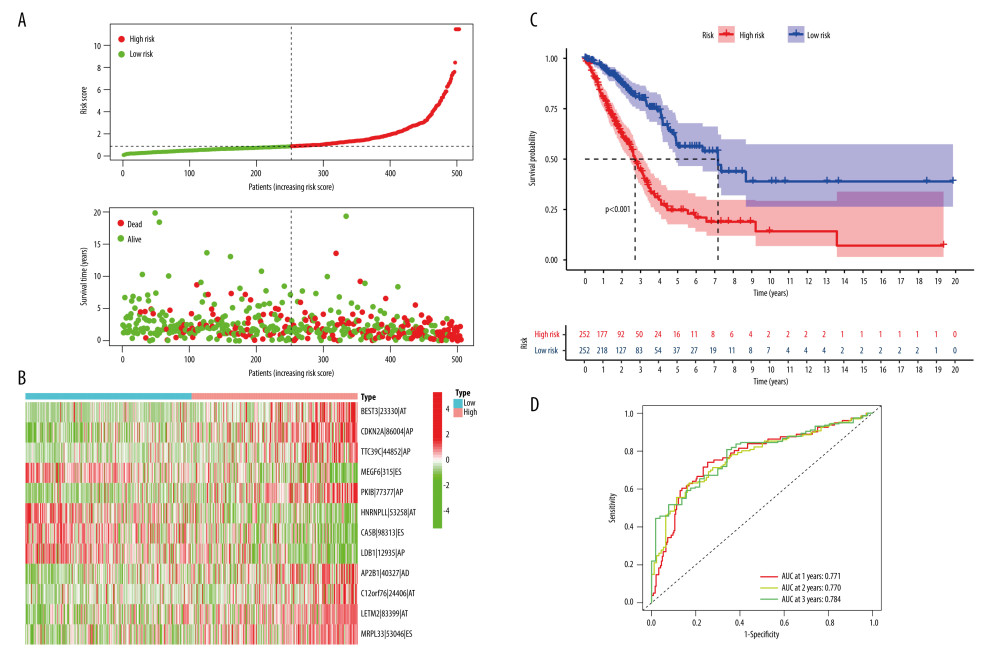 Figure 3. Validation of the risk model. (A) Patients ranked according to risk scores and distribution curve of risk scores. (B) AS events in the risk subgroup of heatmap. (C) Survival analysis of the risk subgroup. (D) ROC prediction curves of LUAD patients at 1, 2, and 3 years. R Software version 4.0.3 (https://www.r-project.org/).
Figure 3. Validation of the risk model. (A) Patients ranked according to risk scores and distribution curve of risk scores. (B) AS events in the risk subgroup of heatmap. (C) Survival analysis of the risk subgroup. (D) ROC prediction curves of LUAD patients at 1, 2, and 3 years. R Software version 4.0.3 (https://www.r-project.org/). 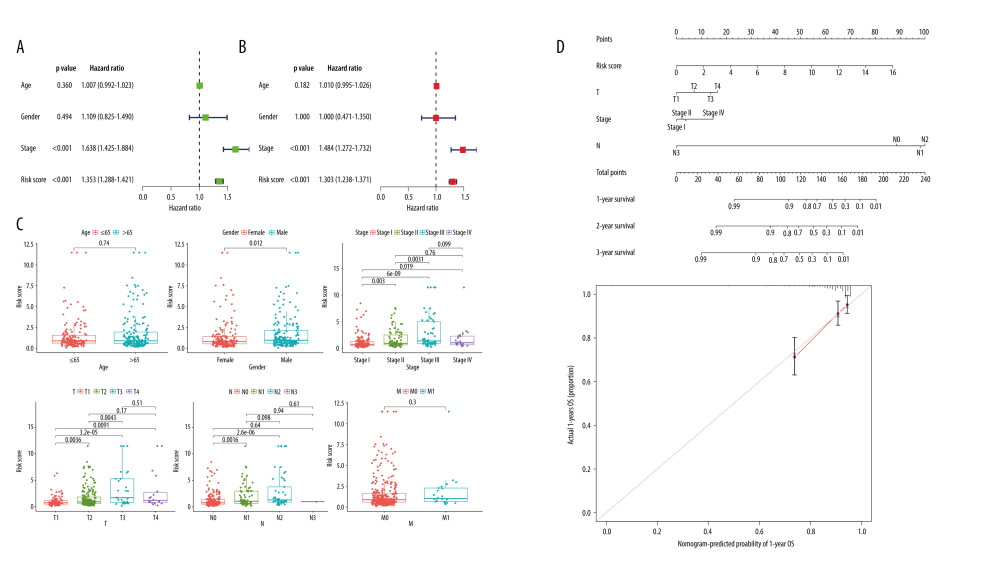 Figure 4. Evaluation of the AS risk model in terms of LUAD prognosis. (A) Univariate Cox regression analysis for clinical characteristics (age, sex, clinical stage) and risk score. (B) Multivariate Cox regression analysis for clinical characteristics (age, sex, clinical stage) and risk score. (C) Difference Analysis between risk subgroup and clinical characteristics. (D) Column line plot and Calibration Chart of the AS-associated risk model. R Software version 4.0.3 (https://www.r-project.org/).
Figure 4. Evaluation of the AS risk model in terms of LUAD prognosis. (A) Univariate Cox regression analysis for clinical characteristics (age, sex, clinical stage) and risk score. (B) Multivariate Cox regression analysis for clinical characteristics (age, sex, clinical stage) and risk score. (C) Difference Analysis between risk subgroup and clinical characteristics. (D) Column line plot and Calibration Chart of the AS-associated risk model. R Software version 4.0.3 (https://www.r-project.org/). 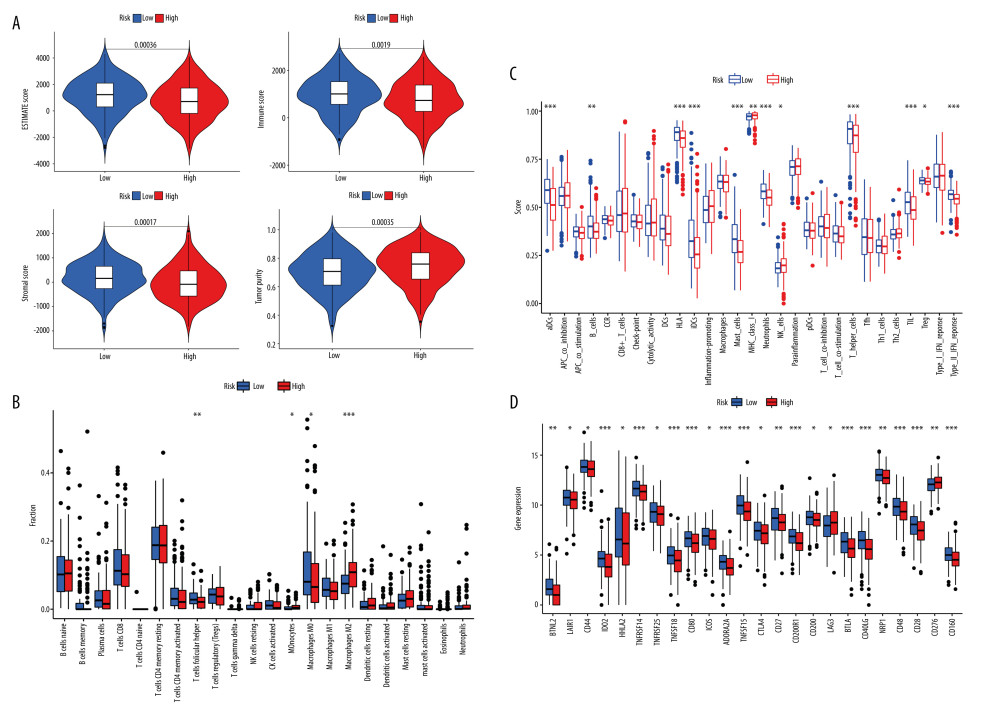 Figure 5. Risk model and immunity. (A) Risk model and tumor microenvironment. (B) Risk model with significantly different immune infiltrating cells. (C) Risk model and 22 types of immune infiltrating cells. (D) Risk model and immune checkpoint. * P<0.05, ** P<0.01, *** P<0.001. R Software version 4.0.3 (https://www.r-project.org/).
Figure 5. Risk model and immunity. (A) Risk model and tumor microenvironment. (B) Risk model with significantly different immune infiltrating cells. (C) Risk model and 22 types of immune infiltrating cells. (D) Risk model and immune checkpoint. * P<0.05, ** P<0.01, *** P<0.001. R Software version 4.0.3 (https://www.r-project.org/). 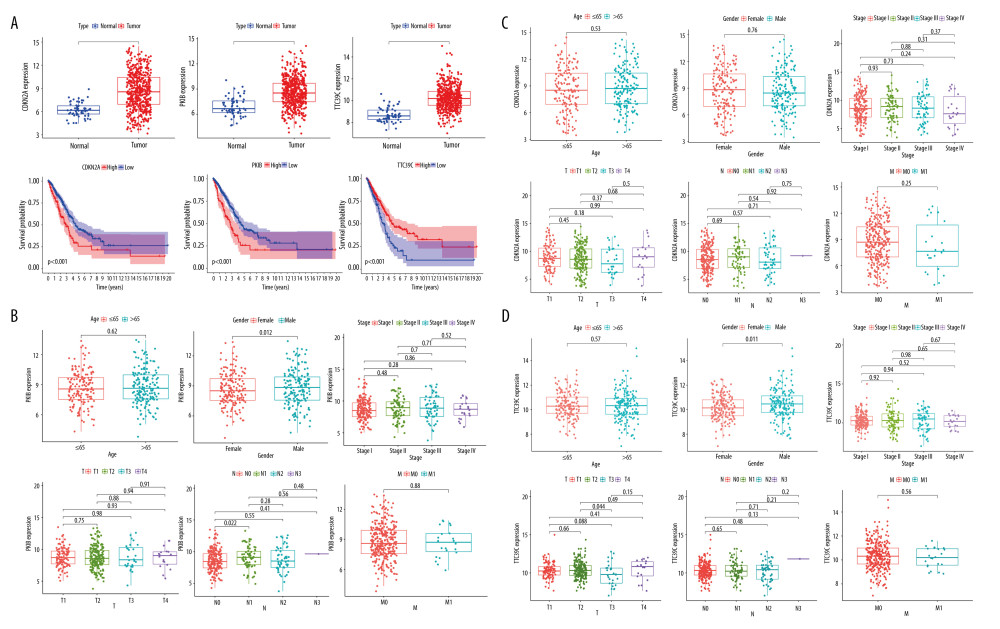 Figure 6. Comprehensive analysis of core genes. (A) Differential expression and survival analysis of CDKN2A, PKIB, and TTC39C among tumor and paraneoplastic tissues. (B) Correlation between PKIB and clinical characteristics. (C) Correlation between TTC29C and clinical characteristics. (D) Correlation between CDKN2A and clinical characteristics. * P<0.05, ** P<0.01, *** P<0.001. R Software version 4.0.3 (https://www.r-project.org/).
Figure 6. Comprehensive analysis of core genes. (A) Differential expression and survival analysis of CDKN2A, PKIB, and TTC39C among tumor and paraneoplastic tissues. (B) Correlation between PKIB and clinical characteristics. (C) Correlation between TTC29C and clinical characteristics. (D) Correlation between CDKN2A and clinical characteristics. * P<0.05, ** P<0.01, *** P<0.001. R Software version 4.0.3 (https://www.r-project.org/). 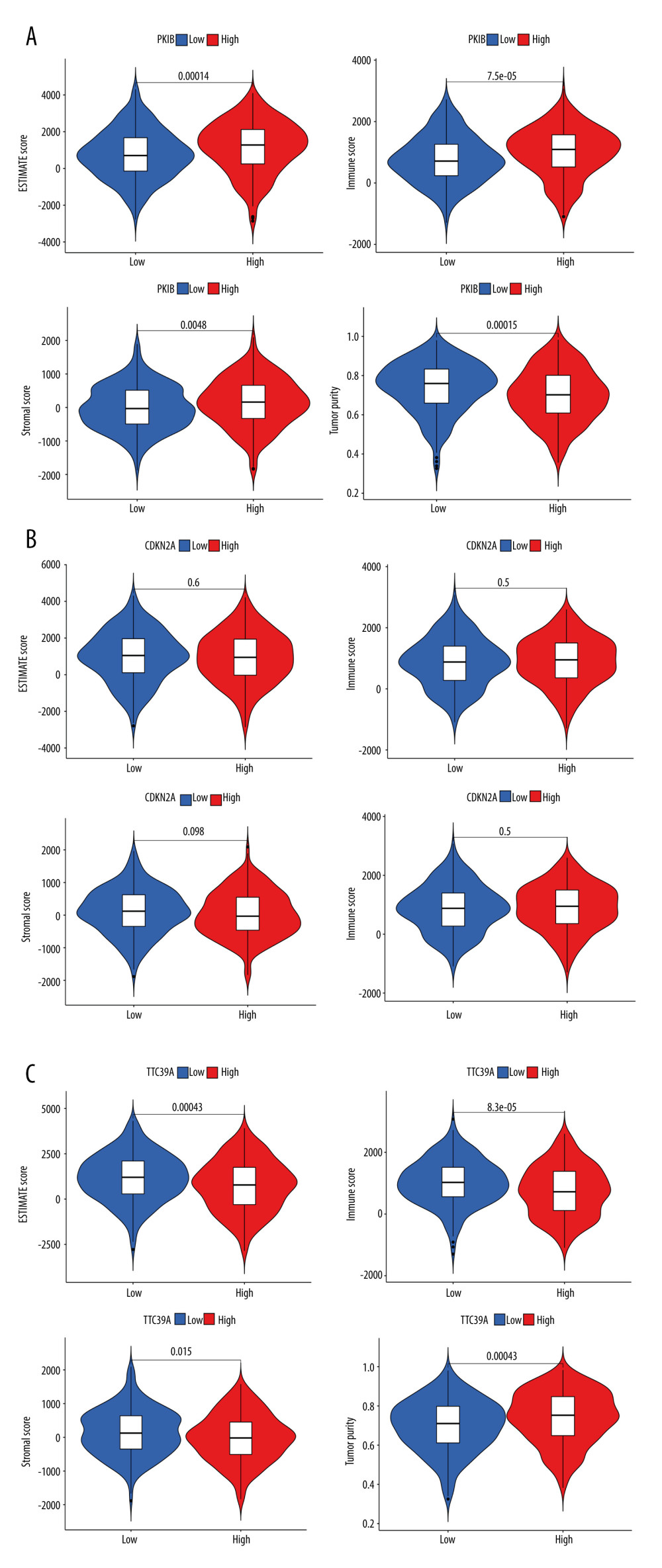 Figure 7. Comprehensive analysis of PKIB, CDKN2A, TTC29C, and tumor microenvironment (TME). (A) PKIB and TME. (B) CDKN2A and TME. (C) TTC39C and TME. R Software version 4.0.3 (https://www.r-project.org/).
Figure 7. Comprehensive analysis of PKIB, CDKN2A, TTC29C, and tumor microenvironment (TME). (A) PKIB and TME. (B) CDKN2A and TME. (C) TTC39C and TME. R Software version 4.0.3 (https://www.r-project.org/). 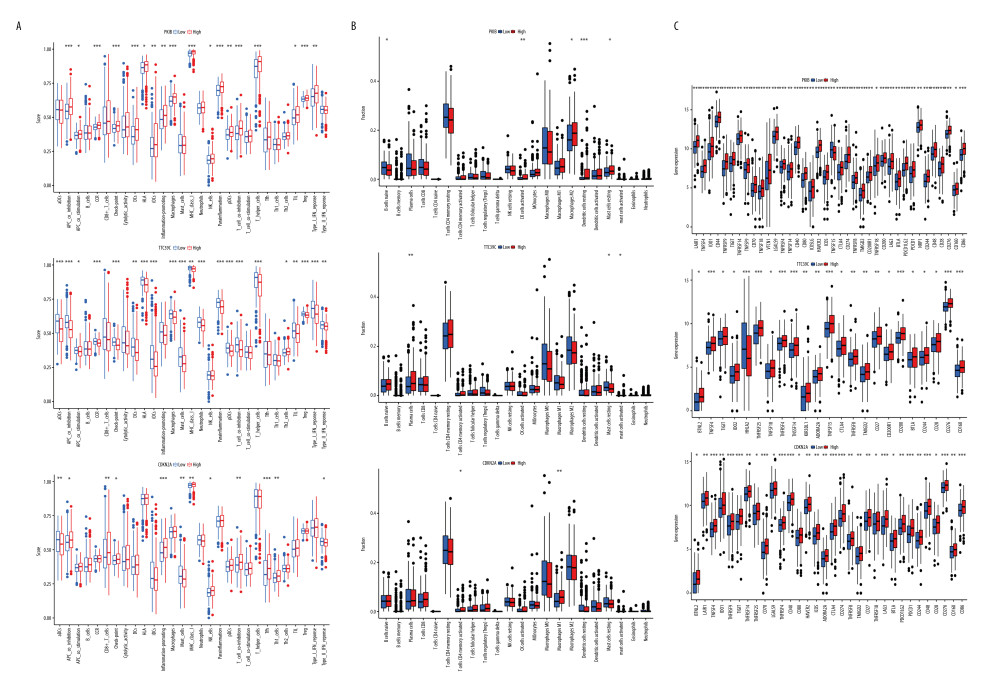 Figure 8. Comprehensive analysis of PKIB, CDKN2A, and TTC29C. (A) PKIB, CDKN2A, TTC29C, and 22 types of immune score. (B) PKIB, CDKN2A, TTC29C, and 22 types of immune infiltrating cells. (C) PKIB, CDKN2A, TTC29C, and immune checkpoint. * P<0.05, ** P<0.01, *** P<0.001. R Software version 4.0.3 (https://www.r-project.org/).
Figure 8. Comprehensive analysis of PKIB, CDKN2A, and TTC29C. (A) PKIB, CDKN2A, TTC29C, and 22 types of immune score. (B) PKIB, CDKN2A, TTC29C, and 22 types of immune infiltrating cells. (C) PKIB, CDKN2A, TTC29C, and immune checkpoint. * P<0.05, ** P<0.01, *** P<0.001. R Software version 4.0.3 (https://www.r-project.org/). 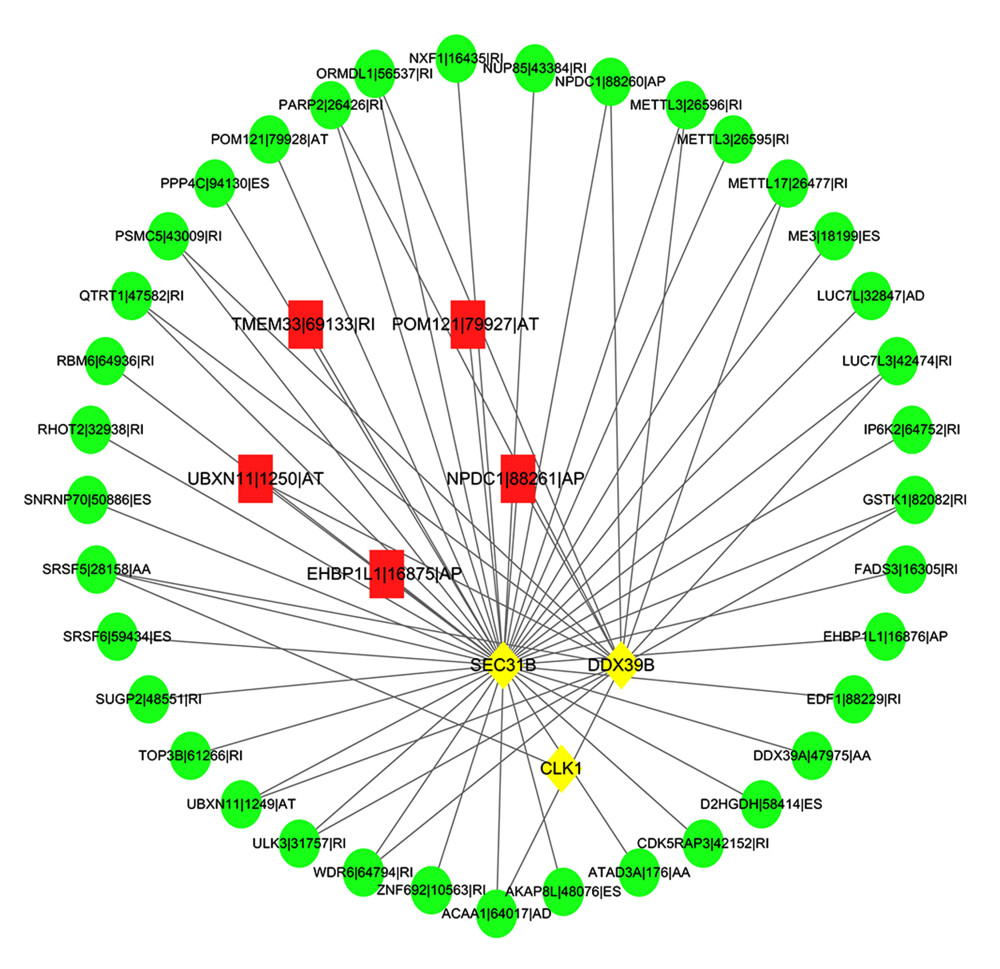 Figure 9. Diagram of the interaction between alternative splicing-alternative splicing factors network. Red represents high-risk AS events, green represents low-risk ASE, and yellow represents alternative splicing factors. Cytoscape version 3.7.2 (https://cytoscape.org/).
Figure 9. Diagram of the interaction between alternative splicing-alternative splicing factors network. Red represents high-risk AS events, green represents low-risk ASE, and yellow represents alternative splicing factors. Cytoscape version 3.7.2 (https://cytoscape.org/). 
References
1. Villalobos P, Wistuba II, Lung cancer biomarkers: Hematol Oncol Clin North Am, 2017; 31(1); 13-29
2. Herbst RS, Morgensztern D, Boshoff C, The biology and management of non-small cell lung cancer: Nature, 2018; 553(7689); 446-54
3. Aoki MN, Amarante MK, de Oliveira CEC, Watanabe MAE, Biomarkers in non-small cell lung cancer: Perspectives of individualized targeted therapy: Anticancer Agents Med Chem, 2018; 18(15); 2070-77
4. Bechara EG, Sebestyen E, Bernardis I, RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation: Mol Cell, 2013; 52(5); 720-33
5. Xu Z, Wei J, Qin F, Hypoxia-associated alternative splicing signature in lung adenocarcinoma: Epigenomics, 2021; 13(1); 47-63
6. Khorrami S, Zavaran Hosseini A, MicroRNA-146a induces immune suppression and drug-resistant colorectal cancer cells: Tumour Biol, 2017; 39(5); 1010428317698365
7. Pillai J, Chincholkar T, Dixit R, Pandey M, A systematic review of proteomic biomarkers in oral squamous cell cancer: World J Surg Oncol, 2021; 19(1); 315
8. Terao T, Kumagi T, Hyodo I, Simple prognostic markers for optimal treatment of patients with unresectable pancreatic cancer: Medicine (Baltimore), 2021; 100(43); e27591
9. Rafei H, El-Bahesh E, Finianos A, Immune-based therapies for non-small cell lung cancer: Anticancer Res, 2017; 37(2); 377-87
10. Shi JY, Bi YY, Yu BF, Alternative splicing events in tumor immune infiltration in colorectal cancer: Front Oncol, 2021; 11; 583547
11. Ryan MC, Cleland J, Kim R, SpliceSeq: A resource for analysis and visualization of RNA-Seq data on alternative splicing and its functional impacts: Bioinformatics, 2012; 28(18); 2385-87
12. Ó Hartaigh B, Gransar H, Callister T, Development and validation of a simple-to-use nomogram for predicting 5-, 10-, and 15-year survival in asymptomatic adults undergoing coronary artery calcium scoring: Cardiovasc Imaging, 2018; 11(3); 450-58
13. Lambrechts D, Wauters E, Boeckx B, Phenotype molding of stromal cells in the lung tumor microenvironment: Nat Med, 2018; 24(8); 1277-89
14. Wood SL, Pernemalm M, Crosbie PA, Whetton AD, The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets: Cancer Treat Rev, 2014; 40(4); 558-66
15. Naylor EC, Desani JK, Chung PK, Targeted therapy and immunotherapy for lung cancer: Surg Oncol Clin N Am, 2016; 25(3); 601-9
16. Cong J, Wang X, Zheng X, Dysfunction of natural killer cells by FBP1-induced inhibition of glycolysis during lung cancer progression: Cell Metab, 2018; 28(2); 243-255e5
17. Conway EM, Pikor LA, Kung SH, Macrophages, inflammation, and lung cancer: Am J Respir Crit Care Med, 2016; 193(2); 116-30
18. Guo X, Zhang Y, Zheng L, Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing: Nat Med, 2018; 24(7); 978-85
19. Wang SS, Liu W, Ly D, Tumor-infiltrating B cells: Their role and application in anti-tumor immunity in lung cancer: Cell Mol Immunol, 2019; 16(1); 6-18
20. Man YG, Stojadinovic A, Mason J, Tumor-infiltrating immune cells promoting tumor invasion and metastasis: Existing theories: J Cancer, 2013; 4(1); 84-95
21. Liu S, Liang J, Liu Z, The role of CD276 in cancers: Front Oncol, 2021; 11; 654684
22. Hayes CS, Labuzan SA, Menke JA, Ttc39c is upregulated during skeletal muscle atrophy and modulates ERK1/2 MAP kinase and hedgehog signaling: J Cell Physiol, 2019; 234(12); 23807-24
23. Meng W, Chan BW, Ezeonwumelu C, A genome-wide association study implicates that the TTC39C gene is associated with diabetic maculopathy with decreased visual acuity: Ophthalmic Genet, 2019; 40(3); 252-58
24. Buchegger K, Silva R, Lopez J, The ERK/MAPK pathway is overexpressed and activated in gallbladder cancer: Pathol Res Pract, 2017; 213(5); 476-82
25. Burotto M, Chiou VL, Lee JM, Kohn EC, The MAPK pathway across different malignancies: A new perspective: Cancer, 2014; 120(22); 3446-56
26. Zou X, Blank M, Targeting p38 MAP kinase signaling in cancer through post-translational modifications: Cancer Lett, 2017; 384; 19-26
27. Dou P, Zhang D, Cheng Z, PKIB promotes cell proliferation and the invasion-metastasis cascade through the PI3K/Akt pathway in NSCLC cells: Exp Biol Med (Maywood), 2016; 241(17); 1911-18
28. Bartsch DK, Sina-Frey M, Lang S, CDKN2A germline mutations in familial pancreatic cancer: Ann Surg, 2002; 236(6); 730-37
29. Song J, Liu J, Lv D, Analysis of genome-wide alternative splicing profiling and development of potential drugs in lung adenocarcinoma: Front Genet, 2021; 12; 767259
30. Li Y, Sun N, Lu Z, Prognostic alternative mRNA splicing signature in non-small cell lung cancer: Cancer Lett, 2017; 393; 40-51
Figures
Figure 1. Flow chart of our study. Visio version 1.5.6.Figure 2. Alternative splicing (AS) of lung adenocarcinoma (LUAD) analysis. (A) UpSet demonstrates the interaction of AS events. (B) UpSet demonstrates the interaction of Prognosis-related AS events. (C) Volcano plot to show prognosis-related AS events. R Software version 4.0.3 (https://www.r-project.org/).Figure 3. Validation of the risk model. (A) Patients ranked according to risk scores and distribution curve of risk scores. (B) AS events in the risk subgroup of heatmap. (C) Survival analysis of the risk subgroup. (D) ROC prediction curves of LUAD patients at 1, 2, and 3 years. R Software version 4.0.3 (https://www.r-project.org/).Figure 4. Evaluation of the AS risk model in terms of LUAD prognosis. (A) Univariate Cox regression analysis for clinical characteristics (age, sex, clinical stage) and risk score. (B) Multivariate Cox regression analysis for clinical characteristics (age, sex, clinical stage) and risk score. (C) Difference Analysis between risk subgroup and clinical characteristics. (D) Column line plot and Calibration Chart of the AS-associated risk model. R Software version 4.0.3 (https://www.r-project.org/).Figure 5. Risk model and immunity. (A) Risk model and tumor microenvironment. (B) Risk model with significantly different immune infiltrating cells. (C) Risk model and 22 types of immune infiltrating cells. (D) Risk model and immune checkpoint. * P<0.05, ** P<0.01, *** P<0.001. R Software version 4.0.3 (https://www.r-project.org/).Figure 6. Comprehensive analysis of core genes. (A) Differential expression and survival analysis of CDKN2A, PKIB, and TTC39C among tumor and paraneoplastic tissues. (B) Correlation between PKIB and clinical characteristics. (C) Correlation between TTC29C and clinical characteristics. (D) Correlation between CDKN2A and clinical characteristics. * P<0.05, ** P<0.01, *** P<0.001. R Software version 4.0.3 (https://www.r-project.org/).Figure 7. Comprehensive analysis of PKIB, CDKN2A, TTC29C, and tumor microenvironment (TME). (A) PKIB and TME. (B) CDKN2A and TME. (C) TTC39C and TME. R Software version 4.0.3 (https://www.r-project.org/).Figure 8. Comprehensive analysis of PKIB, CDKN2A, and TTC29C. (A) PKIB, CDKN2A, TTC29C, and 22 types of immune score. (B) PKIB, CDKN2A, TTC29C, and 22 types of immune infiltrating cells. (C) PKIB, CDKN2A, TTC29C, and immune checkpoint. * P<0.05, ** P<0.01, *** P<0.001. R Software version 4.0.3 (https://www.r-project.org/).Figure 9. Diagram of the interaction between alternative splicing-alternative splicing factors network. Red represents high-risk AS events, green represents low-risk ASE, and yellow represents alternative splicing factors. Cytoscape version 3.7.2 (https://cytoscape.org/). In Press
05 Mar 2024 : Clinical Research
Effects of Thermal Insulation on Recovery and Comfort of Patients Undergoing Holmium Laser LithotripsyMed Sci Monit In Press; DOI: 10.12659/MSM.942836
05 Mar 2024 : Clinical Research
Role of Critical Shoulder Angle in Degenerative Type Rotator Cuff Tears: A Turkish Cohort StudyMed Sci Monit In Press; DOI: 10.12659/MSM.943703
06 Mar 2024 : Clinical Research
Comparison of Outcomes between Single-Level and Double-Level Corpectomy in Thoracolumbar Reconstruction: A ...Med Sci Monit In Press; DOI: 10.12659/MSM.943797
21 Mar 2024 : Meta-Analysis
Economic Evaluation of COVID-19 Screening Tests and Surveillance Strategies in Low-Income, Middle-Income, a...Med Sci Monit In Press; DOI: 10.12659/MSM.943863
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952